You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5822)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Mercruiser Service Manual #14 Alpha I Gen II Outdrives 1991-NewerDocument715 pagesMercruiser Service Manual #14 Alpha I Gen II Outdrives 1991-NewerM5Melo100% (10)
- vxBjICCrQ7eQYyAgq0O3cg - Debit Credit Rules ActivityDocument1 pagevxBjICCrQ7eQYyAgq0O3cg - Debit Credit Rules ActivityAlok PatilNo ratings yet
- Electrical QuantitiesDocument34 pagesElectrical QuantitiesMika VaughnNo ratings yet
- Faculty of Commerce Bachelor of CommerceDocument28 pagesFaculty of Commerce Bachelor of CommercemawandeNo ratings yet
- Brigada Eskwela Regional Kick-Off Program FINAL PDFDocument2 pagesBrigada Eskwela Regional Kick-Off Program FINAL PDFmsantos2870% (10)
- Introduction To The Gas LawsDocument5 pagesIntroduction To The Gas LawsMika VaughnNo ratings yet
- Factors Affecting The Rate of Chemical Reactions: Before You ReadDocument9 pagesFactors Affecting The Rate of Chemical Reactions: Before You ReadMika VaughnNo ratings yet
- Balancing Redox Reactions by The Ion-Electron Method AcidDocument3 pagesBalancing Redox Reactions by The Ion-Electron Method AcidMika VaughnNo ratings yet
- Cathodic and Anodic ProtectionDocument2 pagesCathodic and Anodic ProtectionMika VaughnNo ratings yet
- POGIL Activity - Collision TheoryDocument4 pagesPOGIL Activity - Collision TheoryMika VaughnNo ratings yet
- Chapter10-Physical Significance of IntegralDocument66 pagesChapter10-Physical Significance of IntegralMika VaughnNo ratings yet
- NonClinical Labs Inspections List (10-1-2000 Through 9-27-2020)Document78 pagesNonClinical Labs Inspections List (10-1-2000 Through 9-27-2020)Mika VaughnNo ratings yet
- Corrosion Behavior Between Dental Implant AbutmentDocument3 pagesCorrosion Behavior Between Dental Implant AbutmentMika VaughnNo ratings yet
- Expt #1 - Molecular Modeling of Alkanes Assigned Reading/Viewing - Lab Manual, #1, Expt 18a. Hyperchem InstallationDocument9 pagesExpt #1 - Molecular Modeling of Alkanes Assigned Reading/Viewing - Lab Manual, #1, Expt 18a. Hyperchem InstallationMika VaughnNo ratings yet
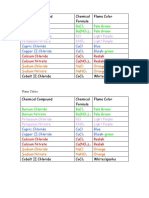
- Flame Colors:: Barium Chloride Bacl Pale Green Barium Nitrate Ba (No) Pale GreenDocument1 pageFlame Colors:: Barium Chloride Bacl Pale Green Barium Nitrate Ba (No) Pale GreenMika VaughnNo ratings yet
- Carib Studies Module 2 NotesDocument96 pagesCarib Studies Module 2 NotesMika Vaughn100% (1)
- BIOL 1263 Living Organisms II Tutorial Questions-Mycology SectionDocument2 pagesBIOL 1263 Living Organisms II Tutorial Questions-Mycology SectionMika VaughnNo ratings yet
- Utilization of Agro-Industrial Wastes For The Production of Quality Oyster MushroomsDocument10 pagesUtilization of Agro-Industrial Wastes For The Production of Quality Oyster MushroomsMhelvene AlfarasNo ratings yet
- Viber For Business - Advertising Solutions-2Document24 pagesViber For Business - Advertising Solutions-2htetarkar022023No ratings yet
- Development of Entrepeneurship and Role of Edii in GujaratDocument7 pagesDevelopment of Entrepeneurship and Role of Edii in GujaratViŠhål PätělNo ratings yet
- Evans SyndromeDocument13 pagesEvans SyndromerizeviNo ratings yet
- CFP Registration Process (Final) - 3Document39 pagesCFP Registration Process (Final) - 3nishiNo ratings yet
- Dial Plan Implementation: Introducing Call RoutingDocument180 pagesDial Plan Implementation: Introducing Call RoutingGuillermo Ex TottiNo ratings yet
- Nkrumah NeocolonialismDocument205 pagesNkrumah Neocolonialismabdelrehman.almNo ratings yet
- Workstations InventoryDocument37 pagesWorkstations InventoryPacificNo ratings yet
- The RC Oscillator CircuitDocument6 pagesThe RC Oscillator CircuitNishanthi BheemanNo ratings yet
- Mini Cex 1 POP CTTDocument10 pagesMini Cex 1 POP CTTZAHRA RIZQIKA ALIYYA SAFITRI -No ratings yet
- Shoppers StopDocument35 pagesShoppers StopAditi JindalNo ratings yet
- Jersey Documentation 1.0.3 User GuideDocument35 pagesJersey Documentation 1.0.3 User Guidenaresh921No ratings yet
- RA 020 Risk Assessment - Risk Assessment - Installation of Cables in Ducts & TrenchesDocument11 pagesRA 020 Risk Assessment - Risk Assessment - Installation of Cables in Ducts & Trenchesthomson100% (2)
- Workbook For Developmental Communications 1 UNIT 6Document14 pagesWorkbook For Developmental Communications 1 UNIT 6soldadodeslealNo ratings yet
- 4 Step Formula For A Killer IntroductionDocument8 pages4 Step Formula For A Killer IntroductionSHARMINE CABUSASNo ratings yet
- Kakhisong Church A2-ModelDocument1 pageKakhisong Church A2-ModelanzaniNo ratings yet
- 2021 WA2 Science (Physics) 3E ModifiedDocument13 pages2021 WA2 Science (Physics) 3E ModifiedfinNo ratings yet
- EnglishFile4e Pre-Intermediate TG PCM Grammar 12BDocument1 pageEnglishFile4e Pre-Intermediate TG PCM Grammar 12BB Mc0% (1)
- Panadent SystemDocument70 pagesPanadent SystemDhananjay GandageNo ratings yet
- Brigada Solicitation and InvitationDocument3 pagesBrigada Solicitation and Invitationguendolyn templadoNo ratings yet
- HH Sheet HANNAHDocument1 pageHH Sheet HANNAHcirohero24No ratings yet
- Mail Delivery SubsystemDocument9 pagesMail Delivery SubsystemLu GuessaNo ratings yet
- On My Honor, I Have Neither Solicited Nor Received Unauthorized Assistance On This AssignmentDocument6 pagesOn My Honor, I Have Neither Solicited Nor Received Unauthorized Assistance On This AssignmentSuresh KumarNo ratings yet
- Kennel Affix Application: Canine Club, Inc. For The Exclusive Use Ofthe Name ..........................................Document1 pageKennel Affix Application: Canine Club, Inc. For The Exclusive Use Ofthe Name ..........................................Dan GotandaNo ratings yet
- Orthodontic Treatment of Mandibular Anterior Crowding: Key Words: Crowding, Malocclusion, Labial FlaringDocument4 pagesOrthodontic Treatment of Mandibular Anterior Crowding: Key Words: Crowding, Malocclusion, Labial FlaringDunia Medisku OnlineShopNo ratings yet
- Presentation: Like/love + Noun or - Ing FormDocument2 pagesPresentation: Like/love + Noun or - Ing FormKaren GimezNo ratings yet