Download as pdf or txt
You might also like
- Machine Learning Business ReportDocument60 pagesMachine Learning Business Reportshorya74% (54)
- Project Submission Machine Learning - Ankit Bhagat - 8th JanDocument36 pagesProject Submission Machine Learning - Ankit Bhagat - 8th Janankitbhagat100% (7)
- Discovery Studio 2.1Document130 pagesDiscovery Studio 2.1dark_geneNo ratings yet
- Bio Info ProjectDocument16 pagesBio Info Project19BTBT037 S.VishaliNo ratings yet
- Abhilash-SWISS MODEL Seminar 2023Document25 pagesAbhilash-SWISS MODEL Seminar 2023abhilashjgNo ratings yet
- Visualize Variant On StructureDocument11 pagesVisualize Variant On StructureVahid MansouriNo ratings yet
- Homology ModelingDocument22 pagesHomology ModelingBasab GhoshNo ratings yet
- Primer 3e Chap3 Case NewDocument8 pagesPrimer 3e Chap3 Case Newl.purwandiipbNo ratings yet
- Cambridge Structural Database System 2010 RELEASE: Registered Charity No 800579Document53 pagesCambridge Structural Database System 2010 RELEASE: Registered Charity No 800579Non Yobuis NessNo ratings yet
- Protein ModellingDocument53 pagesProtein ModellingiluvgermieNo ratings yet
- Cell Designer Ex 10Document2 pagesCell Designer Ex 10thamizh5550% (1)
- Beldiceanu12a-Model ADocument17 pagesBeldiceanu12a-Model ASophia RoseNo ratings yet
- Protein Modelling: (Building 3D Models of Proteins)Document19 pagesProtein Modelling: (Building 3D Models of Proteins)rednriNo ratings yet
- Lab Report 1 BME 310Document7 pagesLab Report 1 BME 310Can MunganNo ratings yet
- Block Modeling & Product Designation: Stratigraphic ModelDocument8 pagesBlock Modeling & Product Designation: Stratigraphic ModelAl-hakimNo ratings yet
- Chapter 3 Case Study Update Using Smartpls 4: July 2023Document9 pagesChapter 3 Case Study Update Using Smartpls 4: July 2023theresiaellen2020No ratings yet
- SURPAC Model FillingDocument13 pagesSURPAC Model FillingDelfidelfi SatuNo ratings yet
- Admeworks Model Builder Standard Edition (Latest Version) Work FlowDocument20 pagesAdmeworks Model Builder Standard Edition (Latest Version) Work FlowYatindra YadavNo ratings yet
- CDO PricingDocument40 pagesCDO Pricingmexicocity78No ratings yet
- Homologymodeling 150123025144 Conversion Gate01Document27 pagesHomologymodeling 150123025144 Conversion Gate01Jhanvi SNo ratings yet
- Surpac Block Model ConceptsDocument6 pagesSurpac Block Model ConceptsMitra MineralNo ratings yet
- Skyline Small Molecule QuantificationDocument28 pagesSkyline Small Molecule QuantificationShahinuzzamanAdaNo ratings yet
- Data Science and Machine Learning Essentials: Lab 4B - Working With Classification ModelsDocument29 pagesData Science and Machine Learning Essentials: Lab 4B - Working With Classification ModelsaussatrisNo ratings yet
- MOE Homlogy Modelling, Building and Evaluating The Protein StructureDocument13 pagesMOE Homlogy Modelling, Building and Evaluating The Protein Structureokta nursantiNo ratings yet
- Project - Machine Learning-Business Report: By: K Ravi Kumar PGP-Data Science and Business Analytics (PGPDSBA.O.MAR23.A)Document38 pagesProject - Machine Learning-Business Report: By: K Ravi Kumar PGP-Data Science and Business Analytics (PGPDSBA.O.MAR23.A)Ravi KotharuNo ratings yet
- C Lust A Lgby Hand Example 2002Document29 pagesC Lust A Lgby Hand Example 2002Márcio MartinsNo ratings yet
- Bioinformatics Notes - 17Bt54: Module - 4Document48 pagesBioinformatics Notes - 17Bt54: Module - 4Samuel PrinceNo ratings yet
- NCBI BLAST Practical 21042022Document7 pagesNCBI BLAST Practical 21042022meliszaNo ratings yet
- Re Comb 2003Document16 pagesRe Comb 2003mustafaNo ratings yet
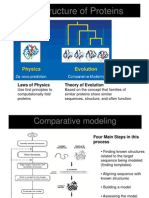
- 3-D Structure of Proteins: Laws of Physics Theory of EvolutionDocument9 pages3-D Structure of Proteins: Laws of Physics Theory of EvolutionladykikyoyunaNo ratings yet
- Homology ModellingDocument21 pagesHomology ModellingJerome FrancisNo ratings yet
- Prottest: Selection of Best-Fit Models of Protein Evolution: Abascal@Mncn - Csic.Es Dposada@Uvigo - Es Rafaz@Mncn - Csic.EsDocument17 pagesProttest: Selection of Best-Fit Models of Protein Evolution: Abascal@Mncn - Csic.Es Dposada@Uvigo - Es Rafaz@Mncn - Csic.EsBodhi DharmaNo ratings yet
- Ebook 038 Tutorial Spss Two Step Cluster Analysis PDFDocument10 pagesEbook 038 Tutorial Spss Two Step Cluster Analysis PDFRex Rieta VillavelezNo ratings yet
- Tools TutorialDocument38 pagesTools TutorialĐỗ Thanh TùngNo ratings yet
- Materials Studio Introductory Tutorial PDFDocument6 pagesMaterials Studio Introductory Tutorial PDFunistarNo ratings yet
- Machine Learning Project: Sneha Sharma PGPDSBA Mar'21 Group 2Document36 pagesMachine Learning Project: Sneha Sharma PGPDSBA Mar'21 Group 2preeti100% (3)
- Machine Learning Project ReportDocument65 pagesMachine Learning Project ReportAbhishek AbhiNo ratings yet
- Tabular Data Generation Can WeDocument12 pagesTabular Data Generation Can WeDhruvesh SojitraNo ratings yet
- 8-Som With E-MinerDocument8 pages8-Som With E-Minertehnic1No ratings yet
- Homolgy ModelingDocument19 pagesHomolgy ModelingJainendra JainNo ratings yet
- Herbs/ Phytochem/ Proteins - HTMLDocument18 pagesHerbs/ Phytochem/ Proteins - HTMLjyoupanishadNo ratings yet
- Need & Emergence of The Field: Speaker Shashi Shekhar Head of Computational Section Biowits Life SciencesDocument59 pagesNeed & Emergence of The Field: Speaker Shashi Shekhar Head of Computational Section Biowits Life SciencesIma kuro-bikeNo ratings yet
- Dali TutorialDocument19 pagesDali TutorialADITI KONARNo ratings yet
- Xlstat Tutorial For ClusteringDocument6 pagesXlstat Tutorial For ClusteringIrina AlexandraNo ratings yet
- Package ABACUS': R Topics DocumentedDocument14 pagesPackage ABACUS': R Topics DocumentedNeilFaverNo ratings yet
- Computer Applications in Chemistry 2 Lec: BY: Sadia HaqDocument27 pagesComputer Applications in Chemistry 2 Lec: BY: Sadia HaqZee ShanNo ratings yet
- Computer Applications 2nd Lec (1) - 2Document27 pagesComputer Applications 2nd Lec (1) - 2Iqra YasmeenNo ratings yet
- Note That There Are Several Different "Basic Blast" Programs Available at Ncbi (Including Nucleotide Blast, Protein Blast, and Blastx)Document10 pagesNote That There Are Several Different "Basic Blast" Programs Available at Ncbi (Including Nucleotide Blast, Protein Blast, and Blastx)Raj Kumar SoniNo ratings yet
- Paper Template Quality2-18Document12 pagesPaper Template Quality2-18Mian HassanNo ratings yet
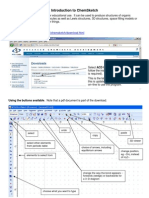
- CHEMSKETCHDocument10 pagesCHEMSKETCHGeliane RochaNo ratings yet
- 2012 Pls-Sem Workshop 5-26-2012 Initial Revised - 2Document25 pages2012 Pls-Sem Workshop 5-26-2012 Initial Revised - 2katonNo ratings yet
- Genetic Algorithm Optimization For Selecting The Best Architecture of A Multi-Layer Perceptron Neural Network: A Credit Scoring CaseDocument8 pagesGenetic Algorithm Optimization For Selecting The Best Architecture of A Multi-Layer Perceptron Neural Network: A Credit Scoring Caseazrim02No ratings yet
- Sww11 Preliminary ReportDocument9 pagesSww11 Preliminary ReportqwertyNo ratings yet
- COMKAT Compartment Model Kinetic Analysis ToolDocument10 pagesCOMKAT Compartment Model Kinetic Analysis ToolIvoPetrovNo ratings yet
- Chapter5: Experiment On Explanation UtilityDocument7 pagesChapter5: Experiment On Explanation UtilityLillian LinNo ratings yet
- PPA-Building Prediction Model MLDocument26 pagesPPA-Building Prediction Model MLDaffa AmmarulNo ratings yet
- What Is Preference Mapping PDFDocument12 pagesWhat Is Preference Mapping PDFRoque VirgilioNo ratings yet
- Process Performance Models: Statistical, Probabilistic & SimulationFrom EverandProcess Performance Models: Statistical, Probabilistic & SimulationNo ratings yet
- Systemic Effects of Surgical Stress:: Dr. H.R.Bhardwaj: Asif AliDocument23 pagesSystemic Effects of Surgical Stress:: Dr. H.R.Bhardwaj: Asif Alimanzoor bhatNo ratings yet
- Topical Vit C and The SkinDocument4 pagesTopical Vit C and The SkinPaulus AnungNo ratings yet
- Cannabis Systematics at The Levels of Family Genus and SpeciesDocument10 pagesCannabis Systematics at The Levels of Family Genus and SpeciesYingjie ZhouNo ratings yet
- ESCHERICHIA COLIaDocument12 pagesESCHERICHIA COLIaEstefania LN VasquezNo ratings yet
- Cellular Structure and FunctionDocument2 pagesCellular Structure and Functionkamalab04No ratings yet
- Senior High School: Learning Module inDocument14 pagesSenior High School: Learning Module inHannahEsparagosaNo ratings yet
- El Sayed2005Document7 pagesEl Sayed2005Romeo Pomarí JuárezNo ratings yet
- Chemical Vs Bio PesticidesDocument34 pagesChemical Vs Bio PesticidesDesh Deepak Sharma60% (5)
- Manual de Plagas y Enfermedades PDFDocument129 pagesManual de Plagas y Enfermedades PDFnil huamaniNo ratings yet
- Pangaea and History of Indonesia IslandDocument42 pagesPangaea and History of Indonesia IslandNila FirmaliaNo ratings yet
- 2 - Prenatal DevelopmentDocument66 pages2 - Prenatal DevelopmentParameswary PerumalNo ratings yet
- Genbio1 Module-1Document31 pagesGenbio1 Module-1Dado Pangan ColagoNo ratings yet
- Cambridge IGCSE Biology (0610) Past Paper Questions and AnswersDocument112 pagesCambridge IGCSE Biology (0610) Past Paper Questions and AnswersKiro XuNo ratings yet
- Cell Junctions NotesDocument3 pagesCell Junctions Notes3DSツTRĪCKSHØTNo ratings yet
- Mito Chloro LectureDocument2 pagesMito Chloro LectureDuaneNo ratings yet
- Is Continued Genetic Improvement of Livestock Sustainable?Document5 pagesIs Continued Genetic Improvement of Livestock Sustainable?leinner vanegasNo ratings yet
- Bacterial Colony Counter: Manual Vs Automatic: Ms. Hemlata Sethi Ms. Sunita YadavDocument3 pagesBacterial Colony Counter: Manual Vs Automatic: Ms. Hemlata Sethi Ms. Sunita YadavIhsanNo ratings yet
- Questionpaper Paper1 June2014Document32 pagesQuestionpaper Paper1 June2014Udoy SahaNo ratings yet
- Chemistry Project 1Document24 pagesChemistry Project 1mahidharNo ratings yet
- MCAS 2019 Released Biology QuestionsDocument25 pagesMCAS 2019 Released Biology QuestionsArFn FNNo ratings yet
- Branches of MicrobiologyDocument2 pagesBranches of MicrobiologyMonika Sharma100% (2)
- Grade 10 Science Sum. TestfinalDocument6 pagesGrade 10 Science Sum. TestfinalHAIDEENo ratings yet
- The Impact of Active and Passive Smoking Upon Health and Neurocognitive Function.Document4 pagesThe Impact of Active and Passive Smoking Upon Health and Neurocognitive Function.AmiraNo ratings yet
- Designer Babies and Genetic Engineering: By: Matea Bagaric, 4mmDocument19 pagesDesigner Babies and Genetic Engineering: By: Matea Bagaric, 4mmzarah jiyavudeenNo ratings yet
- Modern Chemistry Chapter 6-9 Homework AnswersDocument5 pagesModern Chemistry Chapter 6-9 Homework Answersafnapvbseurfgy100% (1)
- Pengaruh Suhu Terhadap Kerja EnzimDocument6 pagesPengaruh Suhu Terhadap Kerja EnzimAlfrilin PadjaoNo ratings yet
- Chapter 1Document40 pagesChapter 1John Lloyd EspirituNo ratings yet
- KT152 GeNeiPure Plasmid ExtractionDocument11 pagesKT152 GeNeiPure Plasmid ExtractionHemant KawalkarNo ratings yet
- Q1 Science 9 Module 3Document31 pagesQ1 Science 9 Module 3apudcrizarcellNo ratings yet
- Lab 5 - 3D Structure ModellingDocument21 pagesLab 5 - 3D Structure ModellingGia HoàngNo ratings yet