Download as pdf or txt
You might also like
- Masterthesis Life CycleDocument83 pagesMasterthesis Life Cycleirfan FENERCİOĞLUNo ratings yet
- Free Cash Manuals - A COMPLETE GUIDE TO PRODUCTION OF PAINTDocument24 pagesFree Cash Manuals - A COMPLETE GUIDE TO PRODUCTION OF PAINTSaidu IsmailaNo ratings yet
- Boards and Beyond PHarmDocument23 pagesBoards and Beyond PHarmstuckinbedNo ratings yet
- EnzymesDocument16 pagesEnzymesMartina ZapataNo ratings yet
- Enzymes atfDocument16 pagesEnzymes atfdowasar398No ratings yet
- Enzymes AtfDocument16 pagesEnzymes AtfHusnaNo ratings yet
- Enzyme InhibitionDocument20 pagesEnzyme InhibitionAnkita guptaNo ratings yet
- BIO307 Lecture 7 (Enzyme Kinetics III)Document11 pagesBIO307 Lecture 7 (Enzyme Kinetics III)Phenyo Mmereki100% (2)
- PDF Pertemuan 3. Enzim Bagian 2Document22 pagesPDF Pertemuan 3. Enzim Bagian 2MnemosyneNo ratings yet
- Properties of Enzyme Inhibition (CH 3, 7)Document18 pagesProperties of Enzyme Inhibition (CH 3, 7)afaf100% (1)
- BasicPharmacologySlides PDFDocument101 pagesBasicPharmacologySlides PDFCabdi WaliNo ratings yet
- Michaelis-Menten Model Michaelis-Menten Model (Cont'd) : - For An Enzyme-Catalyzed ReactionDocument2 pagesMichaelis-Menten Model Michaelis-Menten Model (Cont'd) : - For An Enzyme-Catalyzed ReactionAciel CruzNo ratings yet
- Enzyme KineticsDocument20 pagesEnzyme KineticsIslam SamirNo ratings yet
- Enzyme Kinetics: Properties of EnzymesDocument5 pagesEnzyme Kinetics: Properties of EnzymespokerotiNo ratings yet
- Enzymekinetics 160123162523Document36 pagesEnzymekinetics 160123162523Rxjviie BodaNo ratings yet
- Line Weaver BurkDocument1 pageLine Weaver Burkdeepika kanojiyaNo ratings yet
- 15 EnzymesDocument14 pages15 EnzymesliamfuentezNo ratings yet
- 3 Enzyme KineticsDocument22 pages3 Enzyme KineticsLee Ping Shin100% (1)
- Chapter 7.7 Abbreviated Lecture NotesDocument27 pagesChapter 7.7 Abbreviated Lecture NotesZach MaxwellNo ratings yet
- 10 Evaluation of Enzyme Kinetic ParameterDocument3 pages10 Evaluation of Enzyme Kinetic ParameterAtthapu ThirupathaiahNo ratings yet
- Chapter 3: Enzymes: Lecturer: Mukta BansalDocument26 pagesChapter 3: Enzymes: Lecturer: Mukta BansalR RaguvaranNo ratings yet
- Enzyme KineticsDocument26 pagesEnzyme KineticsLyra LasangreNo ratings yet
- Enzyme KineticsDocument56 pagesEnzyme KineticsP SaranyaNo ratings yet
- 7-Bioc431 Enzymes Kinetics S15Document34 pages7-Bioc431 Enzymes Kinetics S15Shariq Mansoor KhanNo ratings yet
- Enzyme KineticsDocument26 pagesEnzyme KineticsLyra LasangreNo ratings yet
- Alczar 2007 CineticaDocument9 pagesAlczar 2007 CineticaAmanara FreitasNo ratings yet
- Enzyme KineticsDocument72 pagesEnzyme Kineticsitokki otoya100% (1)
- En ZymologyDocument24 pagesEn Zymologywyclife akong'oNo ratings yet
- Assignment #2 - Self-Learning SlidesDocument4 pagesAssignment #2 - Self-Learning Slidesbebble boppleNo ratings yet
- Chapter8&9 Enzyme KineticsDocument68 pagesChapter8&9 Enzyme KineticsSyuhadah NoordinNo ratings yet
- Kjdfks FDocument11 pagesKjdfks FTee BeeNo ratings yet
- Kinetics PDFDocument5 pagesKinetics PDFnewmexicoomfsNo ratings yet
- ECE412 - 3 - AM and SSB ModulationDocument16 pagesECE412 - 3 - AM and SSB ModulationMartine JimenezNo ratings yet
- Lecture Notes On BCH 409: Advanced Enzymology (3 Units) : Enzymes and Life ProcessesDocument21 pagesLecture Notes On BCH 409: Advanced Enzymology (3 Units) : Enzymes and Life ProcessesAkash AroraNo ratings yet
- Presentation On BiochemDocument12 pagesPresentation On BiochemRabia HussainNo ratings yet
- MRQ - #3 Enzyme Kinetics - 2015Document44 pagesMRQ - #3 Enzyme Kinetics - 2015yanuar setiawanNo ratings yet
- Determination KM Dan VmaxDocument9 pagesDetermination KM Dan VmaxKoay Kian YewNo ratings yet
- Derivations OF: Enzyme Kinetics (I)Document20 pagesDerivations OF: Enzyme Kinetics (I)Sabari Krishnan B B100% (1)
- Enzyme NotesDocument18 pagesEnzyme NotesDBPNo ratings yet
- Enzyme Class2Document34 pagesEnzyme Class2Ersin KarataşNo ratings yet
- Exerciselist3 Eja2022 Physics SolutionsDocument4 pagesExerciselist3 Eja2022 Physics SolutionsfelicioNo ratings yet
- Formulario ImprimirDocument9 pagesFormulario Imprimirgiljacobo.13No ratings yet
- Chem452 Quiz 2-KeyDocument6 pagesChem452 Quiz 2-KeyDawlat SalamaNo ratings yet
- 10 Enzymatic ReactionDocument30 pages10 Enzymatic ReactionWidya Nur RamadhaniNo ratings yet
- Lecture On EnzymesDocument48 pagesLecture On EnzymesMary Jean DiazNo ratings yet
- Week3 EnzymeKineticDocument4 pagesWeek3 EnzymeKineticOczhinvia DwitasariNo ratings yet
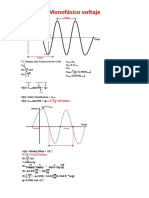
- Monofásico Voltaje: T Tiempo Que Transcurre Un Ciclo F W W V V V 2 V V V 0.707 (V) V 0.637 (V)Document2 pagesMonofásico Voltaje: T Tiempo Que Transcurre Un Ciclo F W W V V V 2 V V V 0.707 (V) V 0.637 (V)Johnny Castro DiazNo ratings yet
- Enzymes L2 Modified For Doctors 2023 2024Document36 pagesEnzymes L2 Modified For Doctors 2023 20249829c7fchsNo ratings yet
- CPII Enzymatic KineticsDocument13 pagesCPII Enzymatic KineticsValentina CretuNo ratings yet
- Enzyme KineticsDocument65 pagesEnzyme KineticsSulabh JainNo ratings yet
- Single Phase Full Wave Controlled Rectifier WithDocument6 pagesSingle Phase Full Wave Controlled Rectifier WithAhmed JamalNo ratings yet
- A DV DT D DTDocument5 pagesA DV DT D DTHOANJO1983No ratings yet
- Introduction To Using The Smith Chart: Bill WilsonDocument2 pagesIntroduction To Using The Smith Chart: Bill Wilsonani18100% (2)
- Chapter 2: Forces and Motion Chapter 2: Forces and Motion: Physics SPM 2018Document13 pagesChapter 2: Forces and Motion Chapter 2: Forces and Motion: Physics SPM 2018The Tech DeveloperNo ratings yet
- Chapter 2 P-2 Enzyme-Inhibition 1Document39 pagesChapter 2 P-2 Enzyme-Inhibition 1Raihan I. SakibNo ratings yet
- How Enzymes Work?Document30 pagesHow Enzymes Work?Nguyễn SunNo ratings yet
- Answer Key Chapter 11Document6 pagesAnswer Key Chapter 11linNo ratings yet
- Sant Longowal Institute of Engineering and Technology: Report On Multisim PracticeDocument17 pagesSant Longowal Institute of Engineering and Technology: Report On Multisim PracticeGurwinder SinghNo ratings yet
- Gas MixtureDocument9 pagesGas MixtureMihai MirceaNo ratings yet
- Ad. BPPK 14Document42 pagesAd. BPPK 14Sachin BagewadiNo ratings yet
- Tffi I Ffi Ffirct,: Qftfitteise BR, YDocument2 pagesTffi I Ffi Ffirct,: Qftfitteise BR, YTawhid ZihadNo ratings yet
- Medicine Periphery RosterDocument5 pagesMedicine Periphery RosterTawhid ZihadNo ratings yet
- CF0 RXBDAw TExDocument5 pagesCF0 RXBDAw TExTawhid ZihadNo ratings yet
- Medicine Is Fun Part 5 - 2020Document37 pagesMedicine Is Fun Part 5 - 2020Tawhid Zihad100% (2)
- Acute and Chronic Kidney DiseaseDocument7 pagesAcute and Chronic Kidney DiseaseTawhid ZihadNo ratings yet
- Renal Reabsorption and SecretionDocument5 pagesRenal Reabsorption and SecretionTawhid ZihadNo ratings yet
- Renal Blood Flow RegulationDocument3 pagesRenal Blood Flow RegulationTawhid ZihadNo ratings yet
- Genesis - Renal System (DR Zihad)Document75 pagesGenesis - Renal System (DR Zihad)Tawhid ZihadNo ratings yet
- Genesis - GIT Physiology (DR Zihad)Document70 pagesGenesis - GIT Physiology (DR Zihad)Tawhid ZihadNo ratings yet
- Antiallergic Drugs, Drugs For Immune SistemDocument38 pagesAntiallergic Drugs, Drugs For Immune SistemTawhid ZihadNo ratings yet
- Renal Anatomy & Physiology: NotesDocument5 pagesRenal Anatomy & Physiology: NotesTawhid ZihadNo ratings yet
- Step On To Paediatrics 4th EditionDocument376 pagesStep On To Paediatrics 4th EditionTawhid Zihad100% (1)
- Renal Blood Flow RegulationDocument3 pagesRenal Blood Flow RegulationTawhid ZihadNo ratings yet
- Step On To Paediatrics 4th EditionDocument376 pagesStep On To Paediatrics 4th EditionTawhid Zihad100% (1)
- RILEM TC 203-RHM Repair Mortars For Historic MasonryDocument13 pagesRILEM TC 203-RHM Repair Mortars For Historic MasonryAngelos IrakleidisNo ratings yet
- ALP-WP-3.08 Rev 1 PDFDocument46 pagesALP-WP-3.08 Rev 1 PDFOliverBrayanNo ratings yet
- Zinc Reduces The Detection of Cocaine, Methamphetamine, and THC by ELISA Urine TestingDocument8 pagesZinc Reduces The Detection of Cocaine, Methamphetamine, and THC by ELISA Urine TestingParvatha SNo ratings yet
- Capstone Final Paper Stem 12c Group5Document126 pagesCapstone Final Paper Stem 12c Group5arboso.trishamaurice.cabralNo ratings yet
- 4 Bonding P1Document19 pages4 Bonding P1mostafa barakatNo ratings yet
- Hybrid Inorganic-Organic Materials by Sol-Gel Processing of Organofunctional Metal AlkoxidesDocument18 pagesHybrid Inorganic-Organic Materials by Sol-Gel Processing of Organofunctional Metal AlkoxidesHéctor MontenegroNo ratings yet
- Chemistry July 2019 STD 12th Science HSC Maharashtra Board Question PaperDocument3 pagesChemistry July 2019 STD 12th Science HSC Maharashtra Board Question PaperGoneNo ratings yet
- Composites C: Open Access: Jamileh Shojaeiarani, Dilpreet S Bajwa, Saptaparni ChandaDocument22 pagesComposites C: Open Access: Jamileh Shojaeiarani, Dilpreet S Bajwa, Saptaparni ChandaTami TomNo ratings yet
- Chapter 21 NeutralizationDocument24 pagesChapter 21 Neutralizationronayme29No ratings yet
- Effect of PH On Non-Enzymatic Browning Reaction DuringDocument8 pagesEffect of PH On Non-Enzymatic Browning Reaction DuringYazwar KotoNo ratings yet
- Care Symbols For Care Instructions On Textile ProductsDocument6 pagesCare Symbols For Care Instructions On Textile ProductsJuanNo ratings yet
- Grease Selection GuideDocument3 pagesGrease Selection GuideDon - BIN95.comNo ratings yet
- Calamba National ComprehensiveDocument9 pagesCalamba National ComprehensiveMarianne gonzalesNo ratings yet
- Types & Uses of Fibcs Flexible Intermediate Bulk Containers: U-Panel BagsDocument5 pagesTypes & Uses of Fibcs Flexible Intermediate Bulk Containers: U-Panel BagsJorwin ButialNo ratings yet
- Robinson Et Al 1993Document14 pagesRobinson Et Al 1993Daniela MWNo ratings yet
- Utilization of Peanut Shells As Sustainable Fillers For Light Weight and Cost-Effective Composite MaterialDocument7 pagesUtilization of Peanut Shells As Sustainable Fillers For Light Weight and Cost-Effective Composite MaterialPierre GanNo ratings yet
- Approval of LPT ProcedureDocument14 pagesApproval of LPT ProcedureimranNo ratings yet
- A Laboratory Report: Protein ExtractionDocument9 pagesA Laboratory Report: Protein ExtractionNadine RebolledoNo ratings yet
- 0001 Section Two: Structure and Written ExpressionDocument4 pages0001 Section Two: Structure and Written ExpressionJunaidi MlbNo ratings yet
- Connective TissuesDocument6 pagesConnective TissuesPenelopeNo ratings yet
- 04 Aldehydes & Ketones Set Test Final EDocument3 pages04 Aldehydes & Ketones Set Test Final EBad boy boyNo ratings yet
- Biological Control 178 (2023) 105145 M. Yousefvand Et AlDocument5 pagesBiological Control 178 (2023) 105145 M. Yousefvand Et AlGenaina CristofoliNo ratings yet
- R633 Refrigeration Cycle Demonstration UnitDocument5 pagesR633 Refrigeration Cycle Demonstration UnitSkati BoyNo ratings yet
- HyperdesmoDocument5 pagesHyperdesmoBryan CruzNo ratings yet
- Reducing Toxic Substances: The Feasibility of Using Banana Fiber and Abacca Fiber As Disposable Facemask. Review Related LiteratureDocument4 pagesReducing Toxic Substances: The Feasibility of Using Banana Fiber and Abacca Fiber As Disposable Facemask. Review Related LiteraturecassseeeyyyNo ratings yet
- Technical Data Sheet ImpResin90Document3 pagesTechnical Data Sheet ImpResin90juaaltaNo ratings yet
- MicroelementeDocument58 pagesMicroelementeDiana ChiscaNo ratings yet
- Comparative GradesDocument9 pagesComparative Gradesabhimanyu.xplastNo ratings yet