Download as pdf or txt
You might also like
- CO2 and Lime Dosage Sea WaterDocument11 pagesCO2 and Lime Dosage Sea WaterNoureddine Merah100% (2)
- Understanding Selectivity of Hard and Soft Metal Cations Within Biological Systems Using The Subvalence ConceptDocument16 pagesUnderstanding Selectivity of Hard and Soft Metal Cations Within Biological Systems Using The Subvalence ConceptAnother Gaming MailNo ratings yet
- Ferrer2021 Article EnergyCalculationsForPotassiumDocument8 pagesFerrer2021 Article EnergyCalculationsForPotassiumSamuelNo ratings yet
- IzomerizareDocument22 pagesIzomerizareTogan CristianNo ratings yet
- Filgueiras 2006Document9 pagesFilgueiras 2006LeonardoNo ratings yet
- Byrne 2017Document11 pagesByrne 2017saeedNo ratings yet
- Calcium Spec o CresolphtaleinDocument4 pagesCalcium Spec o CresolphtaleinHuỳnh LinhNo ratings yet
- ACS Energy Lett 2016 1 1149Document5 pagesACS Energy Lett 2016 1 1149preemeeNo ratings yet
- Bi 9630647Document22 pagesBi 9630647MsMistiqueNo ratings yet
- Ab Initio: Alkali-Metal-Induced Enhancement of Hydrogen Adsorption in C Fullerene: An StudyDocument7 pagesAb Initio: Alkali-Metal-Induced Enhancement of Hydrogen Adsorption in C Fullerene: An StudyAfsal S ShajahanNo ratings yet
- Role of Water Model On Ion Dissociation at Ambient ConditionsDocument14 pagesRole of Water Model On Ion Dissociation at Ambient ConditionsProficient WritersNo ratings yet
- 1) Potassium Carbonate Phase in Estuarine Sediments: Composition, Formation and Chemical ReactivityDocument8 pages1) Potassium Carbonate Phase in Estuarine Sediments: Composition, Formation and Chemical Reactivitymark_idananNo ratings yet
- Zeta-Potential Study of Calcium Silicate Hydrates Interacting With Alkaline CationsDocument9 pagesZeta-Potential Study of Calcium Silicate Hydrates Interacting With Alkaline CationsPíís DGNo ratings yet
- Stomatal Openings Lab 3 Bio 316Document9 pagesStomatal Openings Lab 3 Bio 316Koketso MogweNo ratings yet
- Modeling of Cesium-137 and Strontium-90 Accumulation in The Freshwater Algae CellsDocument6 pagesModeling of Cesium-137 and Strontium-90 Accumulation in The Freshwater Algae CellsfemalefaustNo ratings yet
- Electrochemical Water Softening: Principle and ApplicationDocument14 pagesElectrochemical Water Softening: Principle and ApplicationThao DophuongNo ratings yet
- Regulating Electrodeposition Morphology in High-Capacity Aluminium and Zinc Battery Anodes Using Interfacial Metal-Substrate BondingDocument13 pagesRegulating Electrodeposition Morphology in High-Capacity Aluminium and Zinc Battery Anodes Using Interfacial Metal-Substrate Bondingpeizx123No ratings yet
- Carbon Energy - 2024 - Shi - Pore Structure and Oxygen Content Design of Amorphous Carbon Toward A Durable Anode ForDocument11 pagesCarbon Energy - 2024 - Shi - Pore Structure and Oxygen Content Design of Amorphous Carbon Toward A Durable Anode ForRaghuNo ratings yet
- 1 s2.0 S0376738818302163 MainDocument8 pages1 s2.0 S0376738818302163 MainLeyla UNo ratings yet
- Aitken - 1983 - T-XCO2 Stability Relations and Phase Equilibria of A Calcic Carbonate ScapoliteDocument12 pagesAitken - 1983 - T-XCO2 Stability Relations and Phase Equilibria of A Calcic Carbonate ScapoliteDinarte JrNo ratings yet
- Organic Reactions Volume 57Document250 pagesOrganic Reactions Volume 57vigogiff1594No ratings yet
- Water Research: Huacheng Xu, Changming Yang, Helong JiangDocument8 pagesWater Research: Huacheng Xu, Changming Yang, Helong JiangResin KusumaNo ratings yet
- Calcium and The Cell /a/all: Commissioned Review ArticleDocument9 pagesCalcium and The Cell /a/all: Commissioned Review ArticleCarlos SánchezNo ratings yet
- 06 PDFDocument26 pages06 PDFJohana Rodriguez RuizNo ratings yet
- Advanced Cooling Tower TreatmentDocument6 pagesAdvanced Cooling Tower TreatmentFawaaz KhurwolahNo ratings yet
- Born-Haber Cycles: Learning ObjectivesDocument2 pagesBorn-Haber Cycles: Learning ObjectivesJoko SusiloNo ratings yet
- Asymmetric Ion-Pairing Catalysis: Katrien Brak and Eric N. JacobsenDocument28 pagesAsymmetric Ion-Pairing Catalysis: Katrien Brak and Eric N. JacobsenGeorgeNo ratings yet
- Growth and Structure of Flocs Following Ele - 2016 - Separation and PurificationDocument7 pagesGrowth and Structure of Flocs Following Ele - 2016 - Separation and PurificationBrett Gonzalez CardenasNo ratings yet
- 10 1016@j Memsci 2018 07Document32 pages10 1016@j Memsci 2018 07Elizabeth ZamoraNo ratings yet
- The Determination of The Ion Selectivity of Synthetic IonDocument9 pagesThe Determination of The Ion Selectivity of Synthetic IonDaniel Loh Ka HengNo ratings yet
- Highly Confined Ions Store Charge More EfficientlyDocument6 pagesHighly Confined Ions Store Charge More EfficientlyFaysal Rahman SunbirNo ratings yet
- Theory of Coprecipitation MethodDocument22 pagesTheory of Coprecipitation MethodSebastian PalaNo ratings yet
- Theoretical Study of The Charge Transport Through C - Based Single-Molecule JunctionsDocument9 pagesTheoretical Study of The Charge Transport Through C - Based Single-Molecule JunctionsalbaquintelaNo ratings yet
- Costentin P Natl Acad Sci Usa Dec 19 2017Document5 pagesCostentin P Natl Acad Sci Usa Dec 19 2017llNo ratings yet
- Physical Water TreatmentDocument10 pagesPhysical Water TreatmentVIHIKA ENGINEERINGNo ratings yet
- Calcium in Biological SystemDocument60 pagesCalcium in Biological SystemM S RahmanNo ratings yet
- Flotacin 2Document40 pagesFlotacin 2Aldo GranadosNo ratings yet
- Acs Inorgchem 6b00930Document3 pagesAcs Inorgchem 6b00930Somnath SenguptaNo ratings yet
- ATOOCV1!3!11 Generation Structure Stability and Reactivity of Carbocations Carbanions Free Radicals Carbenes and NitrenesDocument32 pagesATOOCV1!3!11 Generation Structure Stability and Reactivity of Carbocations Carbanions Free Radicals Carbenes and NitrenesUgan KumarNo ratings yet
- Ion Exchange ThesisDocument6 pagesIon Exchange Thesiskriscundiffevansville100% (2)
- Chem 18.1 Experiment 9 'Ion Exchange ChromatographyDocument6 pagesChem 18.1 Experiment 9 'Ion Exchange ChromatographyNat DabuétNo ratings yet
- Advanced Energy Materials - 2021 - Zhang - Sauna Activation Toward Intrinsic Lattice Deficiency in Carbon NanotubeDocument9 pagesAdvanced Energy Materials - 2021 - Zhang - Sauna Activation Toward Intrinsic Lattice Deficiency in Carbon NanotubeDiani MuhandiramNo ratings yet
- Lipid Structure and Membranes, Summary IDocument3 pagesLipid Structure and Membranes, Summary INorman Albon100% (2)
- Physical Organic Approach To Persistent, Cyclable, Low-Potential Electrolytes For Flow Battery ApplicationsDocument4 pagesPhysical Organic Approach To Persistent, Cyclable, Low-Potential Electrolytes For Flow Battery ApplicationsAli TunaNo ratings yet
- Tutorial 3.4Document6 pagesTutorial 3.4arunabhmNo ratings yet
- Jurnal Kimdas2 p6Document8 pagesJurnal Kimdas2 p6Susilo HadiNo ratings yet
- Physical Characteristics of Calcium Induced K-Carrageenan NetworksDocument6 pagesPhysical Characteristics of Calcium Induced K-Carrageenan NetworksFrancisco100% (1)
- Thema Bachelor Thesis LebenslaufDocument7 pagesThema Bachelor Thesis Lebenslaufafhbfqccp100% (2)
- J Mater ChemDocument13 pagesJ Mater Chemsourav13205No ratings yet
- Sarazen 2017 - Stability of Bound Species During Alkene Reactions On Solid AcidsDocument9 pagesSarazen 2017 - Stability of Bound Species During Alkene Reactions On Solid AcidsDébora FernandesNo ratings yet
- Official URLDocument12 pagesOfficial URLHân TrầnNo ratings yet
- Wang 2012Document7 pagesWang 2012venket sNo ratings yet
- Hodgkin 1952Document45 pagesHodgkin 1952Thalia JuarezNo ratings yet
- Example PDFDocument14 pagesExample PDFJuan PerezNo ratings yet
- Nikelhaksasianoferrat PDFDocument5 pagesNikelhaksasianoferrat PDFWiwin Dwi JayantiNo ratings yet
- IPTC 10693 Recent Advances in Carbonate Stimulation: Fig. 1-Normalized and Averaged Reactivity of CarbonatesDocument8 pagesIPTC 10693 Recent Advances in Carbonate Stimulation: Fig. 1-Normalized and Averaged Reactivity of CarbonatesJose Miguel GonzalezNo ratings yet
- 1 s2.0 S2211285523006468 MainDocument15 pages1 s2.0 S2211285523006468 MainAndre Navarro de MirandaNo ratings yet
- Marcelli 2010Document4 pagesMarcelli 2010Angélica Andrea SalinasNo ratings yet
- Combet 2005Document14 pagesCombet 2005brouuorbNo ratings yet
- Polymerization of Heterocycles (Ring Opening): International Union of Pure and Applied ChemistryFrom EverandPolymerization of Heterocycles (Ring Opening): International Union of Pure and Applied ChemistryS. PenczekNo ratings yet
- Pumps, Channels and Transporters: Methods of Functional AnalysisFrom EverandPumps, Channels and Transporters: Methods of Functional AnalysisNo ratings yet
- TDH Calculation Models Compared 1667648114Document4 pagesTDH Calculation Models Compared 1667648114Saeed AbdNo ratings yet
- Case Study ESPCP PMMDocument2 pagesCase Study ESPCP PMMSaeed AbdNo ratings yet
- Molecular Dynamics Simulations of Surface Tensions of Aqueous Electrolytic SolutionsDocument8 pagesMolecular Dynamics Simulations of Surface Tensions of Aqueous Electrolytic SolutionsSaeed AbdNo ratings yet
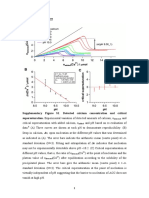
- Supplementary Figures: Detected AddedDocument12 pagesSupplementary Figures: Detected AddedSaeed AbdNo ratings yet
- Molecular Dynamics Simulation On Evaporation of Water and Aqueous Droplets in The Presence of Electric FieldDocument11 pagesMolecular Dynamics Simulation On Evaporation of Water and Aqueous Droplets in The Presence of Electric FieldSaeed AbdNo ratings yet
- Rapid Evaporation of Water On Graphene/Graphene-Oxide: A Molecular Dynamics StudyDocument9 pagesRapid Evaporation of Water On Graphene/Graphene-Oxide: A Molecular Dynamics StudySaeed AbdNo ratings yet
- Instructions To Authors-2017-12-19Document3 pagesInstructions To Authors-2017-12-19Saeed AbdNo ratings yet
- Tuning The Stability of Caco Crystals With Magnesium Ions For The Formation of Aragonite Thin Films On Organic Polymer TemplatesDocument8 pagesTuning The Stability of Caco Crystals With Magnesium Ions For The Formation of Aragonite Thin Films On Organic Polymer TemplatesSaeed AbdNo ratings yet
- Ithenticate - 96444975.docx: 31 04-DEC-2022 02:02PM 93618226Document3 pagesIthenticate - 96444975.docx: 31 04-DEC-2022 02:02PM 93618226Saeed AbdNo ratings yet
- E Ffects of Magnesium Ions and Water Molecules On The Structure of Amorphous Calcium Carbonate: A Molecular Dynamics StudyDocument8 pagesE Ffects of Magnesium Ions and Water Molecules On The Structure of Amorphous Calcium Carbonate: A Molecular Dynamics StudySaeed AbdNo ratings yet
- A Shell Model For The Simulation of Rhombohedral Carbonate Minerals and Their Point DefectsDocument8 pagesA Shell Model For The Simulation of Rhombohedral Carbonate Minerals and Their Point DefectsSaeed AbdNo ratings yet
- Molecular Dynamics Study of Water Evaporation Enhancement Through A Capillary Graphene Bilayer With Tunable HydrophilicityDocument10 pagesMolecular Dynamics Study of Water Evaporation Enhancement Through A Capillary Graphene Bilayer With Tunable HydrophilicitySaeed AbdNo ratings yet
- Acs JCTC 1c00791Document12 pagesAcs JCTC 1c00791Saeed AbdNo ratings yet
- Implementation of Elastic-Plastic Model in PdlammpsDocument7 pagesImplementation of Elastic-Plastic Model in PdlammpsSaeed AbdNo ratings yet
- Colvars ManualDocument71 pagesColvars ManualSaeed AbdNo ratings yet
- A Force Field of Li+, Na+, K+, Mg2+, Ca2+, CL, and in Aqueous Solution Based On The TIP4P/2005 Water Model and Scaled Charges For The IonsDocument17 pagesA Force Field of Li+, Na+, K+, Mg2+, Ca2+, CL, and in Aqueous Solution Based On The TIP4P/2005 Water Model and Scaled Charges For The IonsSaeed AbdNo ratings yet
- Colloids and Surfaces A: Physicochemical and Engineering AspectsDocument9 pagesColloids and Surfaces A: Physicochemical and Engineering AspectsSaeed AbdNo ratings yet
- Implementation of Linear Viscoelasticity Model in PdlammpsDocument7 pagesImplementation of Linear Viscoelasticity Model in PdlammpsSaeed AbdNo ratings yet
- PDLAMMPS - Made Easy: 1 Peridynamic Theory of SolidsDocument8 pagesPDLAMMPS - Made Easy: 1 Peridynamic Theory of SolidsSaeed AbdNo ratings yet
- Water Based EOR by Wettability Alteration in DolomiteDocument8 pagesWater Based EOR by Wettability Alteration in DolomiteSaeed AbdNo ratings yet
- Kspace Extra InfoDocument4 pagesKspace Extra InfoSaeed AbdNo ratings yet
- Communications: Matthew A. Brown, Alok Goel, and Zareen AbbasDocument5 pagesCommunications: Matthew A. Brown, Alok Goel, and Zareen AbbasSaeed AbdNo ratings yet
- LAMMPS Developer GuideDocument14 pagesLAMMPS Developer GuideSaeed AbdNo ratings yet
- IPSXE 2017 Update 8 Release Notes L WDocument19 pagesIPSXE 2017 Update 8 Release Notes L WSaeed AbdNo ratings yet
- Ezthermo: Ezt01: Basic OperationsDocument20 pagesEzthermo: Ezt01: Basic OperationsSaeed AbdNo ratings yet
- Intel® Parallel Studio XE 2017 Update 5: Installation Guide For Linux OSDocument14 pagesIntel® Parallel Studio XE 2017 Update 5: Installation Guide For Linux OSSaeed AbdNo ratings yet
- Phase Envelope Plotting: EzthermoDocument9 pagesPhase Envelope Plotting: EzthermoSaeed AbdNo ratings yet
- Mathcad Vle Eos Svna7Document7 pagesMathcad Vle Eos Svna7Saeed AbdNo ratings yet
- Evaluation of Agricultural LimestoneDocument2 pagesEvaluation of Agricultural LimestonesoumyaNo ratings yet
- Sedimentary and Igneous RocksDocument39 pagesSedimentary and Igneous RocksgengkapakNo ratings yet
- Water SopDocument39 pagesWater SopRohini GadhaweNo ratings yet
- Acid Base and Salt QuestionsDocument3 pagesAcid Base and Salt QuestionsBikash DasNo ratings yet
- Tums Lab ReportDocument2 pagesTums Lab ReportRuby RichiezNo ratings yet
- Soil Stabilization ASSIGNMENTDocument31 pagesSoil Stabilization ASSIGNMENTvishnu_2008No ratings yet
- Parte 2 Mec TerDocument9 pagesParte 2 Mec TerJHONIER ANDRES TORRES URREANo ratings yet
- Geomorphology and Its Different Parameters in The Flood Prone Lower Gandak PainDocument12 pagesGeomorphology and Its Different Parameters in The Flood Prone Lower Gandak PainIJAR JOURNALNo ratings yet
- The S-Block ElementsDocument41 pagesThe S-Block ElementsRavinder singh100% (2)
- The Marine Fish and Invert Reef Aquarium - Book1Document190 pagesThe Marine Fish and Invert Reef Aquarium - Book1Adrian ChiraNo ratings yet
- Climate ChangeDocument124 pagesClimate ChangeNewdeersciNo ratings yet
- Tecnotest 2.3 AggregatesDocument16 pagesTecnotest 2.3 AggregatesLeón SuárezNo ratings yet
- Water Supply Engineering NotesDocument19 pagesWater Supply Engineering NotesAshraf Hussain BelimNo ratings yet
- Qualitative Analysis To Identify Ions!: by Emma Khazzam! 11 Grade, Chemistry!Document18 pagesQualitative Analysis To Identify Ions!: by Emma Khazzam! 11 Grade, Chemistry!emmaekNo ratings yet
- Water and Its Treatment Lecture NotesDocument9 pagesWater and Its Treatment Lecture NotesRose Belle A. GarciaNo ratings yet
- Final Paper Darwin 2.1Document62 pagesFinal Paper Darwin 2.1Raymund Arcos100% (1)
- Material 3382 MCQSDocument283 pagesMaterial 3382 MCQSseven inchesNo ratings yet
- Roll of PH and Double Sulphitation Technique Used in Sugar Industry Sugar CaneDocument55 pagesRoll of PH and Double Sulphitation Technique Used in Sugar Industry Sugar CaneNaresh KumarNo ratings yet
- Properties of Metakaolin Based Geopolymer Incorporating Calcium Carbonate (Aboulayt-2017)Document9 pagesProperties of Metakaolin Based Geopolymer Incorporating Calcium Carbonate (Aboulayt-2017)juan diazNo ratings yet
- Calcium Carbonate-Carbonic Acid EquilibriumDocument15 pagesCalcium Carbonate-Carbonic Acid EquilibriumgombossandorNo ratings yet
- Some Basic Concepts of ChemistryDocument12 pagesSome Basic Concepts of ChemistryNikhil BhattNo ratings yet
- Baroid Completion ManualDocument164 pagesBaroid Completion Manuallatnrythmz100% (1)
- Ammonium Sulfate (ZA)Document8 pagesAmmonium Sulfate (ZA)Ulfa Nurul AuliaNo ratings yet
- TP1 GENIE SEPARATION Version 2 Avec Pressure ExchangerDocument17 pagesTP1 GENIE SEPARATION Version 2 Avec Pressure ExchangerMahran BchatniaNo ratings yet
- Echanisms of Soil-Ime Stabilization: An Interpretive ReviewDocument20 pagesEchanisms of Soil-Ime Stabilization: An Interpretive ReviewArswandya IslamiNo ratings yet
- Thermochemical Energy Storage For A Lunar BaseDocument8 pagesThermochemical Energy Storage For A Lunar BaseXar CrystalNo ratings yet
- High Physical Chemistry PDFDocument300 pagesHigh Physical Chemistry PDFÍris CorreiaNo ratings yet
- Master Thesis Marnix de JongDocument94 pagesMaster Thesis Marnix de JongShashi Ranjan100% (1)
- Dyeability of Polyester and Polyamide Fabrics Employing Citric AcidDocument14 pagesDyeability of Polyester and Polyamide Fabrics Employing Citric AcidAmi SaNo ratings yet