Download as pdf or txt
You might also like
- Metatonin & Pineal GlandDocument107 pagesMetatonin & Pineal GlandSoman100% (2)
- Hydroponics A Modern Technology Supporting The Application of Integrated Crop Management in Greenhouse PDFDocument7 pagesHydroponics A Modern Technology Supporting The Application of Integrated Crop Management in Greenhouse PDFshfgh100% (1)
- Predicting User Interaction On Social Media Using Machine LearninDocument76 pagesPredicting User Interaction On Social Media Using Machine LearninmartinNo ratings yet
- Prediction of Graduation Delay Based On Student CharacterDocument64 pagesPrediction of Graduation Delay Based On Student CharacterAldi Rizki N SNo ratings yet
- Niu Teng 2016 ThesisDocument94 pagesNiu Teng 2016 ThesisJyotirmoy RoyNo ratings yet
- Research 2Document48 pagesResearch 2ruadoanjeanette08No ratings yet
- Preview: Comparison of Machine Learning Algorithms and Their Ensembles For Botnet DetectionDocument11 pagesPreview: Comparison of Machine Learning Algorithms and Their Ensembles For Botnet DetectionChaima BelhediNo ratings yet
- FulltextDocument123 pagesFulltextAAYUSH VARSHNEYNo ratings yet
- 2020 Tong LM PhilDocument97 pages2020 Tong LM PhilShakir NaaimyNo ratings yet
- Rapport PfeDocument82 pagesRapport PfeSarah JedlaouiNo ratings yet
- DissertationDocument137 pagesDissertationyassine badouNo ratings yet
- Chapter 1 5 RESEARCH0328Document76 pagesChapter 1 5 RESEARCH0328Ray CollamatNo ratings yet
- DissertaçãoDocument92 pagesDissertaçãoInes DiasNo ratings yet
- PDF DatastreamDocument51 pagesPDF Datastreamlacan021No ratings yet
- Shi (2023)Document176 pagesShi (2023)Rodrigo RamírezNo ratings yet
- Improving Students Academic Performance With AI ADocument67 pagesImproving Students Academic Performance With AI AdksjahnaviNo ratings yet
- Predicting Clinical Outcomes Via Machine Learning On Electronic Health Records - ThesisDocument60 pagesPredicting Clinical Outcomes Via Machine Learning On Electronic Health Records - ThesisShivin SrivastavaNo ratings yet
- One Class Text Classification Using An Ensemble of ClassifiersDocument71 pagesOne Class Text Classification Using An Ensemble of Classifiersmonitor.whatsapp.quaestNo ratings yet
- Donepudi Naveen Thesis 2023Document54 pagesDonepudi Naveen Thesis 2023The Arena Of GAMERSNo ratings yet
- Ghotra Master ThesisDocument137 pagesGhotra Master ThesisJaved KhanNo ratings yet
- A Reactive Behavioural Model For Context-Aware Semantic DevicesDocument362 pagesA Reactive Behavioural Model For Context-Aware Semantic DevicesIñaki VazquezNo ratings yet
- A Methodology For Detecting Credit Card FraudDocument60 pagesA Methodology For Detecting Credit Card FraudLEM tvNo ratings yet
- Project Proposal 260 CopyDocument38 pagesProject Proposal 260 CopyBorshonNo ratings yet
- MaiDAO M1 Internship ReportDocument51 pagesMaiDAO M1 Internship ReportMai ĐàoNo ratings yet
- ReportDocument55 pagesReportBT20CS028 [AVI]No ratings yet
- Gautam Unr 0139D 13925Document132 pagesGautam Unr 0139D 13925amruth00_210774197No ratings yet
- Thesis Trinh KhoiDocument110 pagesThesis Trinh KhoiQuang Bảo NguyễnNo ratings yet
- Status of Ict in Secondary School ProposalDocument21 pagesStatus of Ict in Secondary School ProposalAdila AhmedNo ratings yet
- (Thesis) Data-Driven Diagnosis in Psychiatry - Wouter Van Der Klift - 5814502 PDFDocument155 pages(Thesis) Data-Driven Diagnosis in Psychiatry - Wouter Van Der Klift - 5814502 PDFJagdishVankarNo ratings yet
- DownloadDocument256 pagesDownloadSURYANo ratings yet
- Deep Learning Enhancement and Privacy-Preserving Deep Learning - ADocument116 pagesDeep Learning Enhancement and Privacy-Preserving Deep Learning - AwildanaulaNo ratings yet
- AmanDocument71 pagesAmansenderNo ratings yet
- BKD-Towards Facilitating Scholarly Communication Using Semantic Technologies-2020Document220 pagesBKD-Towards Facilitating Scholarly Communication Using Semantic Technologies-2020ezoomingNo ratings yet
- Thesis Report Final BU CseDocument34 pagesThesis Report Final BU CseTradNo ratings yet
- Diabetes Disease Prediction Using A Web Tool With The Help of A Machine Learning Model.Document43 pagesDiabetes Disease Prediction Using A Web Tool With The Help of A Machine Learning Model.Iann ClarkeNo ratings yet
- Ascertaining Polarity of Public Opinions On Bangladesh Cricket Through Sentiment AnalysisDocument51 pagesAscertaining Polarity of Public Opinions On Bangladesh Cricket Through Sentiment AnalysisArman AbdullahNo ratings yet
- Budd S 2022 PHD ThesisDocument214 pagesBudd S 2022 PHD ThesisCuuzonNo ratings yet
- Thesis Anum AfzalDocument127 pagesThesis Anum AfzalVivek SahuNo ratings yet
- Advances in Data Science: Methodologies and Applications: Gloria Phillips-Wren Anna Esposito Lakhmi C. Jain EditorsDocument342 pagesAdvances in Data Science: Methodologies and Applications: Gloria Phillips-Wren Anna Esposito Lakhmi C. Jain EditorsIsha DixitNo ratings yet
- Kamphorst Thesis FinalDocument68 pagesKamphorst Thesis FinalarafamgNo ratings yet
- Challenges in Climate Change Communication On Social MediaDocument138 pagesChallenges in Climate Change Communication On Social MediaLasma Ida Mikha Theresia Rogate SitorusNo ratings yet
- Balahmadi ThesisDocument261 pagesBalahmadi ThesisMelvin SpekNo ratings yet
- UC Berkeley Electronic Theses and DissertationsDocument93 pagesUC Berkeley Electronic Theses and DissertationsAnkit ShawNo ratings yet
- LoraDocument51 pagesLoraOMAR CASTILLO CASTULONo ratings yet
- Thesis Without SigDocument54 pagesThesis Without Sigvenneti kiranNo ratings yet
- Raymond - Kassy - 202105 - MSC - EDA Signal ProcessingDocument141 pagesRaymond - Kassy - 202105 - MSC - EDA Signal Processingsuparna.veeturiNo ratings yet
- A Machine Learning Framework For Oncological Margin DetectionDocument108 pagesA Machine Learning Framework For Oncological Margin Detectionmariam askarNo ratings yet
- Teachers - Knowledge Attitudes and Practices Towards The ConductDocument94 pagesTeachers - Knowledge Attitudes and Practices Towards The ConductjanecryztaliegreyesNo ratings yet
- Ibrahim Neji PDFDocument77 pagesIbrahim Neji PDFOneLove EthiopiaNo ratings yet
- Predicting Students Performance by Learning AnalyticsDocument51 pagesPredicting Students Performance by Learning AnalyticsHiếu Nguyễn ChíNo ratings yet
- 2016ipg-011 FRDocument39 pages2016ipg-011 FRFrost ByteNo ratings yet
- FULLTEXT01Document61 pagesFULLTEXT01Arnav RastogiNo ratings yet
- Ashwath Thesis PDFDocument90 pagesAshwath Thesis PDFAnuj MehtaNo ratings yet
- Nanseera Cocis BistDocument95 pagesNanseera Cocis BistMayank PatilNo ratings yet
- Digital Multimedia Forensics and Anti-Forensics - Signals andDocument203 pagesDigital Multimedia Forensics and Anti-Forensics - Signals andlucas caicedoNo ratings yet
- Tagging Application For Customer Data in Withdrawal Form Using XBRL FormatDocument145 pagesTagging Application For Customer Data in Withdrawal Form Using XBRL FormatBio AbidzarNo ratings yet
- A Parent Child Password ManagerDocument106 pagesA Parent Child Password ManagerSowaiba KhanNo ratings yet
- Dissertation PDFDocument92 pagesDissertation PDFMaanikya SharmaNo ratings yet
- Multi-Domain Adaptation For Image Classification Depth EstimatioDocument92 pagesMulti-Domain Adaptation For Image Classification Depth EstimatioЕлена ГрищенкоNo ratings yet
- Kinyua - A Study of Factors That Influence Poor Performance in Mathematics in Secondary Schools by Female Students in Nairobi PDFDocument51 pagesKinyua - A Study of Factors That Influence Poor Performance in Mathematics in Secondary Schools by Female Students in Nairobi PDFAbegail del Castillo100% (1)
- Oscar Munoz GarciaDocument316 pagesOscar Munoz GarciaZoei YoonNo ratings yet
- Bioinformatics Group - Thesis Projects: Last Updated: Nov 19, 2021Document47 pagesBioinformatics Group - Thesis Projects: Last Updated: Nov 19, 2021Joynul Abedin ZubairNo ratings yet
- Deep Learning in Bioinformatics: Introduction, Application, and Perspective in Big Data EraDocument31 pagesDeep Learning in Bioinformatics: Introduction, Application, and Perspective in Big Data EraJoynul Abedin ZubairNo ratings yet
- Supervised Machine Learning Techniques in Bioinformatics: Protein ClassificationDocument109 pagesSupervised Machine Learning Techniques in Bioinformatics: Protein ClassificationJoynul Abedin ZubairNo ratings yet
- Cit 425-Automata Theory, Computability and Formal LanguagesDocument34 pagesCit 425-Automata Theory, Computability and Formal LanguagesJoynul Abedin ZubairNo ratings yet
- Section 01Document5 pagesSection 01Joynul Abedin ZubairNo ratings yet
- Real-Time Driver Drowsiness Detection Using Computer Vision: Mahek Jain, Bhavya Bhagerathi, Sowmyarani C NDocument5 pagesReal-Time Driver Drowsiness Detection Using Computer Vision: Mahek Jain, Bhavya Bhagerathi, Sowmyarani C NJoynul Abedin ZubairNo ratings yet
- Name: Asifuddin Syed Assignment: Homework 2 Course: COSC 5315 Foundations of Computer ScienceDocument8 pagesName: Asifuddin Syed Assignment: Homework 2 Course: COSC 5315 Foundations of Computer ScienceJoynul Abedin ZubairNo ratings yet
- Elisa Troubleshooting TipsDocument3 pagesElisa Troubleshooting TipsDiogenes DezenNo ratings yet
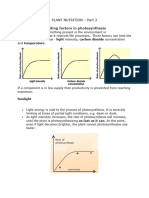
- PLANT NUTRITION Part 2Document10 pagesPLANT NUTRITION Part 2dineo kabasiaNo ratings yet
- Solomon Cb09 PPT 02Document32 pagesSolomon Cb09 PPT 02garimadhamija02No ratings yet
- Chapter 1 - The Nervous SystemDocument8 pagesChapter 1 - The Nervous SystemRadha RamineniNo ratings yet
- Ahar Parinamkar BhavDocument9 pagesAhar Parinamkar BhavDr Prajakta BNo ratings yet
- The Cell CycleDocument72 pagesThe Cell CycleJerry Jeroum RegudoNo ratings yet
- 3 Intravenous-TherapyDocument4 pages3 Intravenous-TherapyMarie Louise Nicole TuvillaNo ratings yet
- 9700 s11 QP 22Document16 pages9700 s11 QP 22Ahmed Kaleem Khan NiaziNo ratings yet
- Free Closed Door Coaching Prof Arconado - Microbiology - 200 Items Key PDFDocument14 pagesFree Closed Door Coaching Prof Arconado - Microbiology - 200 Items Key PDFAnne MorenoNo ratings yet
- SIPDocument20 pagesSIPLuiiiNo ratings yet
- Agri 101: Reflection Report On Pest Management and DiseasesDocument4 pagesAgri 101: Reflection Report On Pest Management and DiseasesIkuzwe Aime PacifiqueNo ratings yet
- 1 Sistem MuskuloskeletalDocument27 pages1 Sistem MuskuloskeletalAshar AbilowoNo ratings yet
- Science Sample Paper Class-8 (2013 - 14) : (1x3 3) I. Very Short Answer QuestionsDocument3 pagesScience Sample Paper Class-8 (2013 - 14) : (1x3 3) I. Very Short Answer QuestionsTechnical GamingNo ratings yet
- Classification of OrganismDocument35 pagesClassification of OrganismJnll JstnNo ratings yet
- Effectiveness of Guava 1Document16 pagesEffectiveness of Guava 1Arellano Reyson100% (1)
- Dapus Anatomi HidungDocument4 pagesDapus Anatomi HidungFadlan HafizhNo ratings yet
- Fundamentals of Bio Informatics Multiple Choice Question (GuruKpo)Document7 pagesFundamentals of Bio Informatics Multiple Choice Question (GuruKpo)GuruKPO100% (1)
- Buongiorno Lineman Manual Hack-Triphasic Supramaximal Compressed Model0Document16 pagesBuongiorno Lineman Manual Hack-Triphasic Supramaximal Compressed Model0YG1No ratings yet
- Journal of Biological RhythmsDocument12 pagesJournal of Biological RhythmsJaved AkhtarNo ratings yet
- ĐỀ TỰ LUYỆN SỐ 2 KEYDocument9 pagesĐỀ TỰ LUYỆN SỐ 2 KEYYinnieNo ratings yet
- COVID-19 ANTIGEN SKYTEST BrochureDocument1 pageCOVID-19 ANTIGEN SKYTEST BrochurerianNo ratings yet
- Ra 7586 - Nipas ActDocument10 pagesRa 7586 - Nipas ActggbadayosNo ratings yet
- Poner en Práctica: Ejercicio 1. Completa Las Oraciones Con oDocument2 pagesPoner en Práctica: Ejercicio 1. Completa Las Oraciones Con oDIEGONo ratings yet
- STEM 5.9D 2.0 Explain STEMscopedia Eng PDFDocument5 pagesSTEM 5.9D 2.0 Explain STEMscopedia Eng PDFspaghett 1No ratings yet
- 2.01 Medical Term PrefixesDocument60 pages2.01 Medical Term PrefixesNicholas FerroniNo ratings yet
- DipladeniaDocument3 pagesDipladeniaCeban DanaNo ratings yet
- De Thi Thu Dai Hoc Mon Tieng Anh Truong THPT Chuyen DH SP Ha Noi de So 1Document7 pagesDe Thi Thu Dai Hoc Mon Tieng Anh Truong THPT Chuyen DH SP Ha Noi de So 1anhlananhNo ratings yet
- BT224 Lec2 Cells OrgansDocument57 pagesBT224 Lec2 Cells OrgansMukul SuryawanshiNo ratings yet