Download as pdf or txt
You might also like
- 0 AlignMeDocument6 pages0 AlignMeCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- s12859 018 2189 Z - Samsa2 PDFDocument11 pagess12859 018 2189 Z - Samsa2 PDFSuran Ravindran NambisanNo ratings yet
- Rover Variant Calling ToolDocument5 pagesRover Variant Calling ToolTung NguyenNo ratings yet
- Multiple Sequence AlignmentDocument6 pagesMultiple Sequence AlignmentGeovani Gregorio Rincon NavasNo ratings yet
- Pieper NucleicAcidsRes 2004Document6 pagesPieper NucleicAcidsRes 2004ctak2022No ratings yet
- Clu StalDocument6 pagesClu StalAna María OchoaNo ratings yet
- 2016 Article 141Document11 pages2016 Article 141Gopal GNo ratings yet
- Praline 3Document18 pagesPraline 3CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Metabolic Network Prediction Through Pairwise Rati - Roche-Lima Et Al. - 2014Document13 pagesMetabolic Network Prediction Through Pairwise Rati - Roche-Lima Et Al. - 2014Eder Efren PerezNo ratings yet
- 2014 Biomed Res Int 348725Document13 pages2014 Biomed Res Int 348725mbrylinskiNo ratings yet
- Comparison and Evaluation of Multiple Sequence Alignment Tools in BininformaticsDocument6 pagesComparison and Evaluation of Multiple Sequence Alignment Tools in Bininformaticsprashant_kaushal_2No ratings yet
- Scansite 20 Proteome-Wide Prediction of Cell SignaDocument8 pagesScansite 20 Proteome-Wide Prediction of Cell SignaAarushi sharmaNo ratings yet
- (Methods in Molecular Biology, 2231) Kazutaka Katoh - Multiple Sequence Alignment - Methods and Protocols-Humana (2020)Document322 pages(Methods in Molecular Biology, 2231) Kazutaka Katoh - Multiple Sequence Alignment - Methods and Protocols-Humana (2020)Diego AlmeidaNo ratings yet
- Unit IiiDocument27 pagesUnit IiiDr. R. K. Selvakesavan PSGRKCWNo ratings yet
- Background:: A Modeling and Simulation Web Tool For Plant BiologistsDocument3 pagesBackground:: A Modeling and Simulation Web Tool For Plant Biologistssk3 khanNo ratings yet
- Sharma 2014Document13 pagesSharma 2014László SágiNo ratings yet
- Multiple Sequence Alignment: Hamid Hamzeiy Izmir Institute of TechnologyDocument6 pagesMultiple Sequence Alignment: Hamid Hamzeiy Izmir Institute of TechnologyHamid HamzeiyNo ratings yet
- Software: Next-Generation Sequence Alignment SoftwareDocument3 pagesSoftware: Next-Generation Sequence Alignment Softwaredj tarpNo ratings yet
- DNA Sequencing: Present Status and Future Challenges: Elaine Mardis Washington University Genome Sequencing CenterDocument26 pagesDNA Sequencing: Present Status and Future Challenges: Elaine Mardis Washington University Genome Sequencing Centerm224550No ratings yet
- Reconfigurable Accelerator For TheDocument46 pagesReconfigurable Accelerator For TheRajasekar PanneerselvamNo ratings yet
- Art - 253A10.1186 - 252Fs13742 015 0064 7Document11 pagesArt - 253A10.1186 - 252Fs13742 015 0064 7Hassan AshrafNo ratings yet
- A Comprehensive Evaluation of Assembly Scaffold ToolsDocument15 pagesA Comprehensive Evaluation of Assembly Scaffold ToolsMarina Célia Nunes Ferreira Da C SilvaNo ratings yet
- 04 Craig JPRDocument9 pages04 Craig JPRTNo ratings yet
- J.D. Opdyke - Permutation Tests (And Sampling Without Replacement) Orders of Magnitude Faster Using SAS - ScribeDocument40 pagesJ.D. Opdyke - Permutation Tests (And Sampling Without Replacement) Orders of Magnitude Faster Using SAS - ScribejdopdykeNo ratings yet
- Roberts Pachter 2013Document16 pagesRoberts Pachter 2013Awawawawa UwuwuwuwuNo ratings yet
- A Benchmark of Batch-Effect Correction Methods For Single-Cell RNA Sequencing DataDocument32 pagesA Benchmark of Batch-Effect Correction Methods For Single-Cell RNA Sequencing DataJKNo ratings yet
- 448 ScalablePM CM 448Document11 pages448 ScalablePM CM 448Ubiquitous Computing and Communication JournalNo ratings yet
- Theory: Sequence Alignment Is A Process of Aligning Two Sequences To Achieve Maximum Levels ofDocument5 pagesTheory: Sequence Alignment Is A Process of Aligning Two Sequences To Achieve Maximum Levels ofGopi ShankarNo ratings yet
- Scalable String Matching Framework Enhanced by Pattern Clustering - Ubiquitous Computing and Communication JournalDocument10 pagesScalable String Matching Framework Enhanced by Pattern Clustering - Ubiquitous Computing and Communication JournalUbiquitous Computing and Communication JournalNo ratings yet
- A Study On Protein Sequence Clustering With OPTICSDocument6 pagesA Study On Protein Sequence Clustering With OPTICSJournal of ComputingNo ratings yet
- Mutscan-A Flexible R Package For Efficient End-To-End Analysis of Multiplexed Assays of Variant Effect DataDocument22 pagesMutscan-A Flexible R Package For Efficient End-To-End Analysis of Multiplexed Assays of Variant Effect DataSergio VillicañaNo ratings yet
- 2015 Article 60Document12 pages2015 Article 60Suany Quesada CalderonNo ratings yet
- 29) Altschul 1997Document14 pages29) Altschul 1997Paulette DlNo ratings yet
- Using Repeatmasker To Identify Repetitive Elements in Genomic SequencesDocument14 pagesUsing Repeatmasker To Identify Repetitive Elements in Genomic SequencesslimcharliNo ratings yet
- Blast 2 S, A New Tool For Comparing Protein and Nucleotide SequencesDocument4 pagesBlast 2 S, A New Tool For Comparing Protein and Nucleotide SequencesXâlmañ KháñNo ratings yet
- PSIPREDDocument8 pagesPSIPREDVaibhavi AwaleNo ratings yet
- Next Generation SequencingDocument70 pagesNext Generation SequencingARUN KUMARNo ratings yet
- Extracted Pages From An Overview of Multiple Sequence AlignmentsDocument6 pagesExtracted Pages From An Overview of Multiple Sequence AlignmentsCristina MendozaNo ratings yet
- Document For CentralizationDocument36 pagesDocument For CentralizationSai GaneshwarNo ratings yet
- Research Article: RECORD: Reference-Assisted Genome Assembly For Closely Related GenomesDocument10 pagesResearch Article: RECORD: Reference-Assisted Genome Assembly For Closely Related GenomesGopal GNo ratings yet
- A Text-Constrained Prosodic System For Speaker VerificationDocument4 pagesA Text-Constrained Prosodic System For Speaker Verificationrasto80No ratings yet
- (73 Finding Protein Similarities With Nucleotide Sequence DatabasesDocument22 pages(73 Finding Protein Similarities With Nucleotide Sequence DatabasesMatias VéjarNo ratings yet
- Sequenceserver: A Modern Graphical User Interface For Custom BLAST DatabasesDocument3 pagesSequenceserver: A Modern Graphical User Interface For Custom BLAST DatabasesywurmNo ratings yet
- Tau Ten Hahn 2012Document5 pagesTau Ten Hahn 2012Shiva Shankar Reddy NimmalaNo ratings yet
- BLASTDocument4 pagesBLASTdia320576No ratings yet
- Scaffolder - Software For Manual Genome Scaffolding: ArticleDocument7 pagesScaffolder - Software For Manual Genome Scaffolding: ArticleAna Carolina MatiussiNo ratings yet
- Fasta Sequence DatabaseDocument17 pagesFasta Sequence DatabaseKlevis XhyraNo ratings yet
- Joint Compressed Sensing and Enhanced Whale OptimiDocument18 pagesJoint Compressed Sensing and Enhanced Whale OptimisurendranathNo ratings yet
- JASPAR: An Open-Access Database For Eukaryotic Transcription Factor Binding Pro®lesDocument4 pagesJASPAR: An Open-Access Database For Eukaryotic Transcription Factor Binding Pro®lesFahad HassanNo ratings yet
- A Distributed Approach To Multi-Objective Evolutionary Generation of Fuzzy Rule-Based Classifiers From Big DataDocument22 pagesA Distributed Approach To Multi-Objective Evolutionary Generation of Fuzzy Rule-Based Classifiers From Big Datajkl316No ratings yet
- Pyro TaggerDocument8 pagesPyro TaggerClaudia MaturanaNo ratings yet
- Next-Generation DNA Sequencing MethodsDocument18 pagesNext-Generation DNA Sequencing MethodsFederico BerraNo ratings yet
- Bioinformatics Is The Inter-Disciplinary Branch of Biology Which Merges Computer Science, Mathematics and Engineering To Study The Biological DataDocument26 pagesBioinformatics Is The Inter-Disciplinary Branch of Biology Which Merges Computer Science, Mathematics and Engineering To Study The Biological DataMalik FhaimNo ratings yet
- Bioinformatics 34 22 3939Document3 pagesBioinformatics 34 22 3939bewieNo ratings yet
- Probability-Based Protein Identification by Searching Sequence Databases Using Mass Spectrometry DataDocument17 pagesProbability-Based Protein Identification by Searching Sequence Databases Using Mass Spectrometry DataEverton MonteiroNo ratings yet
- Fast Bitwise Pattern-Matching Algorithm For DNA Sequences On Modern HardwareDocument13 pagesFast Bitwise Pattern-Matching Algorithm For DNA Sequences On Modern HardwareAnisa Tanzil HasibuanNo ratings yet
- 04-Alinemiento Múltiple de SecuenciasDocument14 pages04-Alinemiento Múltiple de SecuenciasAlexander Ccahuana IgnacioNo ratings yet
- Graph-Based Clustering and Characterization of Repetitive Sequences in Next-Generation Sequencing DataDocument12 pagesGraph-Based Clustering and Characterization of Repetitive Sequences in Next-Generation Sequencing DataFrontiersNo ratings yet
- Next Generation Sequencing and Sequence Assembly: Methodologies and AlgorithmsFrom EverandNext Generation Sequencing and Sequence Assembly: Methodologies and AlgorithmsNo ratings yet
- Energy Management in Wireless Sensor NetworksFrom EverandEnergy Management in Wireless Sensor NetworksRating: 4 out of 5 stars4/5 (1)
- 0 AlignMeDocument6 pages0 AlignMeCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- New Alignment Strategy For TransmembraneDocument9 pagesNew Alignment Strategy For TransmembraneCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Diapositiva 2012tm-Coffee-130709120840-Phpapp02Document24 pagesDiapositiva 2012tm-Coffee-130709120840-Phpapp02CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- On The Accuracy of Homology Modeling and Sequence AlignmentDocument10 pagesOn The Accuracy of Homology Modeling and Sequence AlignmentCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- BALIBASE7REFDocument4 pagesBALIBASE7REFCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- MP-T Improving Membrane Protein Alignment For StructureDocument8 pagesMP-T Improving Membrane Protein Alignment For StructureCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Survey, Comparison and Evaluation of Cross Platform Mobile Application Development ToolsDocument6 pagesSurvey, Comparison and Evaluation of Cross Platform Mobile Application Development ToolsCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Stam 2004Document12 pagesStam 2004CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Praline 3Document18 pagesPraline 3CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Transmembrane Protein Alignment and Fold Recognition Based On Predicted TopologyDocument9 pagesTransmembrane Protein Alignment and Fold Recognition Based On Predicted TopologyCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Bioinformatics: PHAT: A Transmembrane-Specific Substitution MatrixDocument7 pagesBioinformatics: PHAT: A Transmembrane-Specific Substitution MatrixCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- SQLLite Database and ListView AdapterDocument19 pagesSQLLite Database and ListView AdapterCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Mobile First As A Best Practice in Web Design: Otto VarrelaDocument41 pagesMobile First As A Best Practice in Web Design: Otto VarrelaCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Python SyllabusDocument7 pagesPython SyllabusAngel ANo ratings yet
- Elasticsearch 7 Quick Start Guide Get Up and Running With The Distributed Search and Analytics Capabilities of Elasticsearch 9781789803327 1789803322Document318 pagesElasticsearch 7 Quick Start Guide Get Up and Running With The Distributed Search and Analytics Capabilities of Elasticsearch 9781789803327 1789803322ZeroCool ZeroNo ratings yet
- 05 Molap RolapDocument57 pages05 Molap RolapSayed Farid QattalyNo ratings yet
- Taleo ArchitectureDocument2 pagesTaleo Architecturesridhar_eeNo ratings yet
- Activation of CDS ViewsDocument2 pagesActivation of CDS Viewsoscar navaNo ratings yet
- Unit 4: Workload Capture and Replay: Week 4: Performance AnalysisDocument11 pagesUnit 4: Workload Capture and Replay: Week 4: Performance Analysissan1507No ratings yet
- Mukesh Vudhayagiri - 4 Year(s) 8 Month(s)Document3 pagesMukesh Vudhayagiri - 4 Year(s) 8 Month(s)Lavi764 BarbieNo ratings yet
- Chapter-7 Introduction To Access 2013Document3 pagesChapter-7 Introduction To Access 2013Ishaan ChatterjeeNo ratings yet
- How To Create Universes Using BEx Queries As Data Sources With SAP BusinessObjects Information Design Tool 4.1Document23 pagesHow To Create Universes Using BEx Queries As Data Sources With SAP BusinessObjects Information Design Tool 4.1john91067% (6)
- Data Structures and AlgorithmsDocument41 pagesData Structures and AlgorithmsMatthew TedunjaiyeNo ratings yet
- Csmodel NotesDocument79 pagesCsmodel NotesbrentsoanNo ratings yet
- Exam: 1Z0-931 1Z0-931-F: NO.1 A. B. C. D. EDocument15 pagesExam: 1Z0-931 1Z0-931-F: NO.1 A. B. C. D. EQuincy JonesNo ratings yet
- CH 08Document33 pagesCH 08Yasser SabriNo ratings yet
- Database Management 1Document10 pagesDatabase Management 1Sadeepa NimashNo ratings yet
- DatawarehouseDocument6 pagesDatawarehouseJaveriaNo ratings yet
- Imperva DBFDocument11 pagesImperva DBFFabricio RamírezNo ratings yet
- Lesson 12 - Parameter and StatisticDocument16 pagesLesson 12 - Parameter and StatisticjenniferNo ratings yet
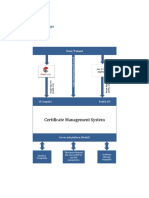
- Certificate ManagerDocument4 pagesCertificate ManagerCWF - AADHYA .ecosecretzNo ratings yet
- Demantra Training Installation InstructionsDocument29 pagesDemantra Training Installation InstructionsAdityaNo ratings yet
- Database Management-1 - Nayyar-ZaidiDocument18 pagesDatabase Management-1 - Nayyar-ZaidiHuyen TranNo ratings yet
- BSIT2E Assignment - Adonis P. SarmientoDocument2 pagesBSIT2E Assignment - Adonis P. SarmientoMark StewartNo ratings yet
- Query ParallelismDocument8 pagesQuery Parallelismbrittain markaleNo ratings yet
- MIS Model WkyDocument23 pagesMIS Model Wkyhabtamu mesfinNo ratings yet
- Benefits, Challenges and Tools of Big Data Management PDFDocument9 pagesBenefits, Challenges and Tools of Big Data Management PDFFrancisco Javier Medina VillarrealNo ratings yet
- Sap Basis FileDocument73 pagesSap Basis FilePulkit JainNo ratings yet
- CH 33Document13 pagesCH 33hiwot ytayewNo ratings yet
- Advances in Data Science and Analytics Concepts and Paradigms - Advances in Data Science and Analytics Concepts and Paradigms (M. Niranjanamurthy, Hemant Kumar Gianey Etc.)Document353 pagesAdvances in Data Science and Analytics Concepts and Paradigms - Advances in Data Science and Analytics Concepts and Paradigms (M. Niranjanamurthy, Hemant Kumar Gianey Etc.)jonathan HerreraNo ratings yet
- Question BankDocument6 pagesQuestion BankManish PingaleNo ratings yet
- Atm System 1Document50 pagesAtm System 1Anjali KumavatNo ratings yet
- Chapter 2Document35 pagesChapter 2Sairam PrakashNo ratings yet