
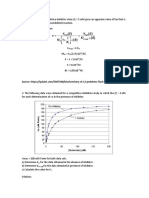
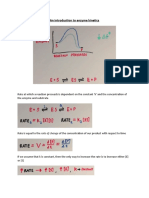
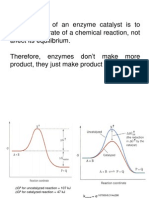
Basic Pharmacology Slides 2020
Basic Pharmacology Slides 2020
You might also like
- A.1. Competitive InhibitionDocument5 pagesA.1. Competitive InhibitionFlorecita Cabañog100% (2)
- SMART Asthma TherapyDocument4 pagesSMART Asthma TherapyKen Won100% (1)
- Enzymes: Jason Ryan, MD, MPHDocument102 pagesEnzymes: Jason Ryan, MD, MPHMohamed AdelNo ratings yet
- Protein IIIDocument18 pagesProtein IIIph.mt.pharmaNo ratings yet
- Enzyme Class2Document34 pagesEnzyme Class2Ersin KarataşNo ratings yet
- 7 Kinetika Enzim InhibitorDocument69 pages7 Kinetika Enzim InhibitorHernanda WidipasuryaNo ratings yet
- Enzyme KineticsDocument20 pagesEnzyme KineticsIslam SamirNo ratings yet
- 5 Enzyme Kinetics-InhibitionDocument40 pages5 Enzyme Kinetics-InhibitionJoel SmolanoffNo ratings yet
- LineweaverBurk PlotDocument17 pagesLineweaverBurk PlotJawadNo ratings yet
- Enzyme Kinetics: Properties of EnzymesDocument5 pagesEnzyme Kinetics: Properties of EnzymespokerotiNo ratings yet
- BCM Enzyme II 200lDocument46 pagesBCM Enzyme II 200lewoozino1234No ratings yet
- Enzymes All NUBDocument8 pagesEnzymes All NUBPIH SHTNo ratings yet
- Chapter 2 P-2 Enzyme-Inhibition 1Document39 pagesChapter 2 P-2 Enzyme-Inhibition 1Raihan I. SakibNo ratings yet
- 153A Final ReviewDocument72 pages153A Final ReviewAswin ArumugamNo ratings yet
- Lab 3 Enzyme KineticsDocument16 pagesLab 3 Enzyme KineticsjojojojoNo ratings yet
- Enzyme KineticsDocument90 pagesEnzyme KineticsAmrit LalNo ratings yet
- Biochemeng Enzyme Kinetics - Additional HandoutDocument22 pagesBiochemeng Enzyme Kinetics - Additional HandoutFlower GNo ratings yet
- Enzyme Kinetics Mechanism and InhibitionDocument38 pagesEnzyme Kinetics Mechanism and InhibitionManoj SigdelNo ratings yet
- Lecture 3: Part 3 Enzyme Kinetics and Inhibition: DR Evelyne DeplazesDocument18 pagesLecture 3: Part 3 Enzyme Kinetics and Inhibition: DR Evelyne Deplazeskristal eliasNo ratings yet
- Boards and Beyond PHarmDocument23 pagesBoards and Beyond PHarmstuckinbedNo ratings yet
- Biology Biomolecules Part 2Document14 pagesBiology Biomolecules Part 2Ralph Kenneth LunaNo ratings yet
- Enzymes All NUBDocument9 pagesEnzymes All NUBPIH SHTNo ratings yet
- Enzymes L2 Modified For Doctors 2023 2024Document36 pagesEnzymes L2 Modified For Doctors 2023 20249829c7fchsNo ratings yet
- Enzyme KineticsDocument56 pagesEnzyme KineticsP SaranyaNo ratings yet
- Enzyme2 2012Document45 pagesEnzyme2 2012Surya Prakash KabiNo ratings yet
- Enzyme InhibitionDocument12 pagesEnzyme Inhibitionderickh72jNo ratings yet
- LEC12 EnzInhib 08Document12 pagesLEC12 EnzInhib 08Megan GohNo ratings yet
- Enzyme Kinetics: Medical Biochemistry, Lecture 24Document39 pagesEnzyme Kinetics: Medical Biochemistry, Lecture 24jeyankarunanithiNo ratings yet
- Chapter 6 - The Behavior of Proteins ENZYMES-2018Document111 pagesChapter 6 - The Behavior of Proteins ENZYMES-2018ruaa mhmadNo ratings yet
- More Enzyme KineticsDocument18 pagesMore Enzyme KineticsKaanNo ratings yet
- EnzymesDocument16 pagesEnzymesMartina ZapataNo ratings yet
- Enzyme Inhibits PlotsDocument24 pagesEnzyme Inhibits Plotsleehmin80No ratings yet
- En ZymologyDocument24 pagesEn Zymologywyclife akong'oNo ratings yet
- Basic Pharmacology Book ColorDocument24 pagesBasic Pharmacology Book ColorTawhid ZihadNo ratings yet
- Enzyme Kinetics - InhibitionDocument40 pagesEnzyme Kinetics - InhibitionRodney Baldwin100% (1)
- How Much Enzyme Have I Got? Enzyme "Activity"Document15 pagesHow Much Enzyme Have I Got? Enzyme "Activity"Julija SabulytėNo ratings yet
- Berenice Ortiz BIB1 Act2 U1Document4 pagesBerenice Ortiz BIB1 Act2 U1Berenice O EscalanteNo ratings yet
- 6 Behavior of Proteins Enzymes PDFDocument35 pages6 Behavior of Proteins Enzymes PDFKelly SisonNo ratings yet
- Enzim Dan KoenzimDocument24 pagesEnzim Dan KoenzimPutri RiduanNo ratings yet
- Enymes L2 2019 NC 6thDocument26 pagesEnymes L2 2019 NC 6thChamara ChathurangaNo ratings yet
- Lecture 7-SOLS 2023Document36 pagesLecture 7-SOLS 2023Arghadeep DasNo ratings yet
- Enzyme NotesDocument18 pagesEnzyme NotesDBPNo ratings yet
- PDF Pertemuan 3. Enzim Bagian 2Document22 pagesPDF Pertemuan 3. Enzim Bagian 2MnemosyneNo ratings yet
- Enzyme, M-I 024Document67 pagesEnzyme, M-I 024daman dhamiNo ratings yet
- 2 Enzymes & Enzyme KineticsDocument37 pages2 Enzymes & Enzyme KineticsNovVie VietTha Sccor IINo ratings yet
- Part 4 Case Study - Industrial EnzymesDocument33 pagesPart 4 Case Study - Industrial EnzymesyahmedpersNo ratings yet
- Catalysis and Enzyme Kinetics PDFDocument43 pagesCatalysis and Enzyme Kinetics PDFBlessings Chawinga100% (1)
- Properties of Enzyme Inhibition (CH 3, 7)Document18 pagesProperties of Enzyme Inhibition (CH 3, 7)afaf100% (1)
- Enzymes AtfDocument16 pagesEnzymes AtfHusnaNo ratings yet
- Enzyme Kinetics Journal 1Document12 pagesEnzyme Kinetics Journal 1Nadia NovitaNo ratings yet
- Dose-Response AtfDocument24 pagesDose-Response AtfHusnaNo ratings yet
- BIO307 Lecture 7 (Enzyme Kinetics III)Document11 pagesBIO307 Lecture 7 (Enzyme Kinetics III)Phenyo Mmereki100% (2)
- Chapter 8 KineticsDocument54 pagesChapter 8 KineticsSanthoshsv 143No ratings yet
- Enzyme KineticsDocument5 pagesEnzyme KineticsCarlo Quinsayas SablanNo ratings yet
- BIO 203 Biochemistry I by Seyhun YURDUGÜL, PH.D.: Enzyme InhibitionDocument59 pagesBIO 203 Biochemistry I by Seyhun YURDUGÜL, PH.D.: Enzyme InhibitionYasin Çağrı KılıçerNo ratings yet
- Materi Kuliah Biokimia Stmi Ke 11 (Enzyme Kinetics)Document72 pagesMateri Kuliah Biokimia Stmi Ke 11 (Enzyme Kinetics)soni_abdullahNo ratings yet
- CHPE422 - Chapter Three - Part 3Document21 pagesCHPE422 - Chapter Three - Part 3KusmakarNo ratings yet
- Lec 7 and 8 (Ch. 5) Enz KinDocument37 pagesLec 7 and 8 (Ch. 5) Enz KinRamy El-HadadNo ratings yet
- 2 Factors Aff & KM, MMeq, DRP 2021Document29 pages2 Factors Aff & KM, MMeq, DRP 2021Srishti GoenkaNo ratings yet
- LEC 07 - Enzyme KineticsDocument36 pagesLEC 07 - Enzyme Kineticseliza makNo ratings yet
- Relationship Between Tonsils and Iga Nephropathy As Well As Indications of TonsillectomyDocument10 pagesRelationship Between Tonsils and Iga Nephropathy As Well As Indications of TonsillectomyCabdi WaliNo ratings yet
- 2024bulletin PDFDocument26 pages2024bulletin PDFCabdi WaliNo ratings yet
- BehavioralSlides PDFDocument196 pagesBehavioralSlides PDFCabdi Wali100% (1)
- PsychSlides PDFDocument434 pagesPsychSlides PDFCabdi WaliNo ratings yet
- BiostatsEpiSlides PDFDocument245 pagesBiostatsEpiSlides PDFCabdi WaliNo ratings yet
- ImmunoSlides PDFDocument424 pagesImmunoSlides PDFCabdi WaliNo ratings yet
- Fluid Therapy Final 2019 DR TaanoDocument39 pagesFluid Therapy Final 2019 DR TaanoCabdi WaliNo ratings yet
- HemeSlides PDFDocument812 pagesHemeSlides PDFCabdi WaliNo ratings yet
- Ecology: of The Disease or Dynamic of Disease Transmission Chapter TwoDocument42 pagesEcology: of The Disease or Dynamic of Disease Transmission Chapter TwoCabdi WaliNo ratings yet
- Acute and Chronic Kidney DiseaseDocument20 pagesAcute and Chronic Kidney DiseaseCabdi WaliNo ratings yet
- Septic Arthritis & OsteomyelitisDocument42 pagesSeptic Arthritis & OsteomyelitisCabdi WaliNo ratings yet
- Common Adult Fractures: Dr. Sayid Omar MohamedDocument15 pagesCommon Adult Fractures: Dr. Sayid Omar MohamedCabdi WaliNo ratings yet
- Ortho History Taking & Physical ExaminationDocument28 pagesOrtho History Taking & Physical ExaminationCabdi WaliNo ratings yet
- Batch6 Clerkship ScheduleDocument1 pageBatch6 Clerkship ScheduleCabdi WaliNo ratings yet
- Daftar Pustaka Makalah ObatDocument2 pagesDaftar Pustaka Makalah Obatnur adiniNo ratings yet
- Intrathecal Antibacterial and Antifungal TherapiesDocument24 pagesIntrathecal Antibacterial and Antifungal TherapiesntnquynhproNo ratings yet
- KAPS Ques - July 2022Document2 pagesKAPS Ques - July 2022majid tariq100% (1)
- AHA Hypertension Guidelines 2017Document22 pagesAHA Hypertension Guidelines 2017Anonymous elSqPhzKNo ratings yet
- Drug Study-Operating RoomDocument6 pagesDrug Study-Operating RoomkathzheinNo ratings yet
- Malone 2013Document8 pagesMalone 2013Nguyễn LongNo ratings yet
- DRUG-STUDY PediaDocument5 pagesDRUG-STUDY PediaRhea Jane Bongcato50% (2)
- School Based Immunization Form Grade 1Document1 pageSchool Based Immunization Form Grade 1maristellaNo ratings yet
- 4 Hospital Medication OrderDocument36 pages4 Hospital Medication Orderसन्दिप क्षेत्रीNo ratings yet
- Hyoscine-N-butylbromide (HNBB)Document1 pageHyoscine-N-butylbromide (HNBB)Kyle DelrosarioNo ratings yet
- Operation FaceBOOKED ArrestsDocument6 pagesOperation FaceBOOKED ArrestsWGN Web Desk0% (1)
- Saba Hanif Presentation...Document19 pagesSaba Hanif Presentation...Saba KhanNo ratings yet
- Common Ophthalmic DrugsDocument3 pagesCommon Ophthalmic DrugsBadr DihamNo ratings yet
- DRUG STUDY 2023 WARD POTASSIUM CHLORIDE AutosavedDocument2 pagesDRUG STUDY 2023 WARD POTASSIUM CHLORIDE AutosavedMary Grace AgataNo ratings yet
- Aceclofenac Vs Diclofenac On Hexosamine LevelsDocument11 pagesAceclofenac Vs Diclofenac On Hexosamine LevelsMartin MoranNo ratings yet
- Research: Legalization of Marijuana in The PhilippinesDocument8 pagesResearch: Legalization of Marijuana in The PhilippinesPhilip Sta. AnaNo ratings yet
- Stannard Chapter5Document22 pagesStannard Chapter5Mae ann BalgoaNo ratings yet
- Print Obat Utk Ruang DokterDocument4 pagesPrint Obat Utk Ruang DokterIntan Kamala AisyiahNo ratings yet
- LECTURE NOTES For Health Science StudentDocument211 pagesLECTURE NOTES For Health Science StudentMohsinNo ratings yet
- Aripiprazole in Meige Syndrome Clinical Response.6Document4 pagesAripiprazole in Meige Syndrome Clinical Response.6tsyrahmaniNo ratings yet
- ChemSex (From A Safety Perspective)Document4 pagesChemSex (From A Safety Perspective)Dominic Milton TrottNo ratings yet
- Drug Study Ferrous Sulfate + FADocument3 pagesDrug Study Ferrous Sulfate + FAKristine ChampnessNo ratings yet
- Cefuroxime 1st PDF TRIAL PDFDocument1 pageCefuroxime 1st PDF TRIAL PDFRyanNo ratings yet
- Answer KeyDocument8 pagesAnswer KeyYap JackyNo ratings yet
- DyslipidemiaDocument11 pagesDyslipidemiaDr-Mohammed ElsawyNo ratings yet
- Pharma Drug StudyDocument56 pagesPharma Drug StudyGrace Pikit Bacsan100% (1)
- Review: Antacids in The Treatment of Gastroduodenal UlcerDocument7 pagesReview: Antacids in The Treatment of Gastroduodenal UlcerClaudia CorjăuceanuNo ratings yet
- YohimbeDocument2 pagesYohimbejssshNo ratings yet
- Montair LC TabletsDocument9 pagesMontair LC Tabletskurutala100% (1)




































































































Download as pdf or txt
You might also like
- A.1. Competitive InhibitionDocument5 pagesA.1. Competitive InhibitionFlorecita Cabañog100% (2)
- SMART Asthma TherapyDocument4 pagesSMART Asthma TherapyKen Won100% (1)
- Enzymes: Jason Ryan, MD, MPHDocument102 pagesEnzymes: Jason Ryan, MD, MPHMohamed AdelNo ratings yet
- Protein IIIDocument18 pagesProtein IIIph.mt.pharmaNo ratings yet
- Enzyme Class2Document34 pagesEnzyme Class2Ersin KarataşNo ratings yet
- 7 Kinetika Enzim InhibitorDocument69 pages7 Kinetika Enzim InhibitorHernanda WidipasuryaNo ratings yet
- Enzyme KineticsDocument20 pagesEnzyme KineticsIslam SamirNo ratings yet
- 5 Enzyme Kinetics-InhibitionDocument40 pages5 Enzyme Kinetics-InhibitionJoel SmolanoffNo ratings yet
- LineweaverBurk PlotDocument17 pagesLineweaverBurk PlotJawadNo ratings yet
- Enzyme Kinetics: Properties of EnzymesDocument5 pagesEnzyme Kinetics: Properties of EnzymespokerotiNo ratings yet
- BCM Enzyme II 200lDocument46 pagesBCM Enzyme II 200lewoozino1234No ratings yet
- Enzymes All NUBDocument8 pagesEnzymes All NUBPIH SHTNo ratings yet
- Chapter 2 P-2 Enzyme-Inhibition 1Document39 pagesChapter 2 P-2 Enzyme-Inhibition 1Raihan I. SakibNo ratings yet
- 153A Final ReviewDocument72 pages153A Final ReviewAswin ArumugamNo ratings yet
- Lab 3 Enzyme KineticsDocument16 pagesLab 3 Enzyme KineticsjojojojoNo ratings yet
- Enzyme KineticsDocument90 pagesEnzyme KineticsAmrit LalNo ratings yet
- Biochemeng Enzyme Kinetics - Additional HandoutDocument22 pagesBiochemeng Enzyme Kinetics - Additional HandoutFlower GNo ratings yet
- Enzyme Kinetics Mechanism and InhibitionDocument38 pagesEnzyme Kinetics Mechanism and InhibitionManoj SigdelNo ratings yet
- Lecture 3: Part 3 Enzyme Kinetics and Inhibition: DR Evelyne DeplazesDocument18 pagesLecture 3: Part 3 Enzyme Kinetics and Inhibition: DR Evelyne Deplazeskristal eliasNo ratings yet
- Boards and Beyond PHarmDocument23 pagesBoards and Beyond PHarmstuckinbedNo ratings yet
- Biology Biomolecules Part 2Document14 pagesBiology Biomolecules Part 2Ralph Kenneth LunaNo ratings yet
- Enzymes All NUBDocument9 pagesEnzymes All NUBPIH SHTNo ratings yet
- Enzymes L2 Modified For Doctors 2023 2024Document36 pagesEnzymes L2 Modified For Doctors 2023 20249829c7fchsNo ratings yet
- Enzyme KineticsDocument56 pagesEnzyme KineticsP SaranyaNo ratings yet
- Enzyme2 2012Document45 pagesEnzyme2 2012Surya Prakash KabiNo ratings yet
- Enzyme InhibitionDocument12 pagesEnzyme Inhibitionderickh72jNo ratings yet
- LEC12 EnzInhib 08Document12 pagesLEC12 EnzInhib 08Megan GohNo ratings yet
- Enzyme Kinetics: Medical Biochemistry, Lecture 24Document39 pagesEnzyme Kinetics: Medical Biochemistry, Lecture 24jeyankarunanithiNo ratings yet
- Chapter 6 - The Behavior of Proteins ENZYMES-2018Document111 pagesChapter 6 - The Behavior of Proteins ENZYMES-2018ruaa mhmadNo ratings yet
- More Enzyme KineticsDocument18 pagesMore Enzyme KineticsKaanNo ratings yet
- EnzymesDocument16 pagesEnzymesMartina ZapataNo ratings yet
- Enzyme Inhibits PlotsDocument24 pagesEnzyme Inhibits Plotsleehmin80No ratings yet
- En ZymologyDocument24 pagesEn Zymologywyclife akong'oNo ratings yet
- Basic Pharmacology Book ColorDocument24 pagesBasic Pharmacology Book ColorTawhid ZihadNo ratings yet
- Enzyme Kinetics - InhibitionDocument40 pagesEnzyme Kinetics - InhibitionRodney Baldwin100% (1)
- How Much Enzyme Have I Got? Enzyme "Activity"Document15 pagesHow Much Enzyme Have I Got? Enzyme "Activity"Julija SabulytėNo ratings yet
- Berenice Ortiz BIB1 Act2 U1Document4 pagesBerenice Ortiz BIB1 Act2 U1Berenice O EscalanteNo ratings yet
- 6 Behavior of Proteins Enzymes PDFDocument35 pages6 Behavior of Proteins Enzymes PDFKelly SisonNo ratings yet
- Enzim Dan KoenzimDocument24 pagesEnzim Dan KoenzimPutri RiduanNo ratings yet
- Enymes L2 2019 NC 6thDocument26 pagesEnymes L2 2019 NC 6thChamara ChathurangaNo ratings yet
- Lecture 7-SOLS 2023Document36 pagesLecture 7-SOLS 2023Arghadeep DasNo ratings yet
- Enzyme NotesDocument18 pagesEnzyme NotesDBPNo ratings yet
- PDF Pertemuan 3. Enzim Bagian 2Document22 pagesPDF Pertemuan 3. Enzim Bagian 2MnemosyneNo ratings yet
- Enzyme, M-I 024Document67 pagesEnzyme, M-I 024daman dhamiNo ratings yet
- 2 Enzymes & Enzyme KineticsDocument37 pages2 Enzymes & Enzyme KineticsNovVie VietTha Sccor IINo ratings yet
- Part 4 Case Study - Industrial EnzymesDocument33 pagesPart 4 Case Study - Industrial EnzymesyahmedpersNo ratings yet
- Catalysis and Enzyme Kinetics PDFDocument43 pagesCatalysis and Enzyme Kinetics PDFBlessings Chawinga100% (1)
- Properties of Enzyme Inhibition (CH 3, 7)Document18 pagesProperties of Enzyme Inhibition (CH 3, 7)afaf100% (1)
- Enzymes AtfDocument16 pagesEnzymes AtfHusnaNo ratings yet
- Enzyme Kinetics Journal 1Document12 pagesEnzyme Kinetics Journal 1Nadia NovitaNo ratings yet
- Dose-Response AtfDocument24 pagesDose-Response AtfHusnaNo ratings yet
- BIO307 Lecture 7 (Enzyme Kinetics III)Document11 pagesBIO307 Lecture 7 (Enzyme Kinetics III)Phenyo Mmereki100% (2)
- Chapter 8 KineticsDocument54 pagesChapter 8 KineticsSanthoshsv 143No ratings yet
- Enzyme KineticsDocument5 pagesEnzyme KineticsCarlo Quinsayas SablanNo ratings yet
- BIO 203 Biochemistry I by Seyhun YURDUGÜL, PH.D.: Enzyme InhibitionDocument59 pagesBIO 203 Biochemistry I by Seyhun YURDUGÜL, PH.D.: Enzyme InhibitionYasin Çağrı KılıçerNo ratings yet
- Materi Kuliah Biokimia Stmi Ke 11 (Enzyme Kinetics)Document72 pagesMateri Kuliah Biokimia Stmi Ke 11 (Enzyme Kinetics)soni_abdullahNo ratings yet
- CHPE422 - Chapter Three - Part 3Document21 pagesCHPE422 - Chapter Three - Part 3KusmakarNo ratings yet
- Lec 7 and 8 (Ch. 5) Enz KinDocument37 pagesLec 7 and 8 (Ch. 5) Enz KinRamy El-HadadNo ratings yet
- 2 Factors Aff & KM, MMeq, DRP 2021Document29 pages2 Factors Aff & KM, MMeq, DRP 2021Srishti GoenkaNo ratings yet
- LEC 07 - Enzyme KineticsDocument36 pagesLEC 07 - Enzyme Kineticseliza makNo ratings yet
- Relationship Between Tonsils and Iga Nephropathy As Well As Indications of TonsillectomyDocument10 pagesRelationship Between Tonsils and Iga Nephropathy As Well As Indications of TonsillectomyCabdi WaliNo ratings yet
- 2024bulletin PDFDocument26 pages2024bulletin PDFCabdi WaliNo ratings yet
- BehavioralSlides PDFDocument196 pagesBehavioralSlides PDFCabdi Wali100% (1)
- PsychSlides PDFDocument434 pagesPsychSlides PDFCabdi WaliNo ratings yet
- BiostatsEpiSlides PDFDocument245 pagesBiostatsEpiSlides PDFCabdi WaliNo ratings yet
- ImmunoSlides PDFDocument424 pagesImmunoSlides PDFCabdi WaliNo ratings yet
- Fluid Therapy Final 2019 DR TaanoDocument39 pagesFluid Therapy Final 2019 DR TaanoCabdi WaliNo ratings yet
- HemeSlides PDFDocument812 pagesHemeSlides PDFCabdi WaliNo ratings yet
- Ecology: of The Disease or Dynamic of Disease Transmission Chapter TwoDocument42 pagesEcology: of The Disease or Dynamic of Disease Transmission Chapter TwoCabdi WaliNo ratings yet
- Acute and Chronic Kidney DiseaseDocument20 pagesAcute and Chronic Kidney DiseaseCabdi WaliNo ratings yet
- Septic Arthritis & OsteomyelitisDocument42 pagesSeptic Arthritis & OsteomyelitisCabdi WaliNo ratings yet
- Common Adult Fractures: Dr. Sayid Omar MohamedDocument15 pagesCommon Adult Fractures: Dr. Sayid Omar MohamedCabdi WaliNo ratings yet
- Ortho History Taking & Physical ExaminationDocument28 pagesOrtho History Taking & Physical ExaminationCabdi WaliNo ratings yet
- Batch6 Clerkship ScheduleDocument1 pageBatch6 Clerkship ScheduleCabdi WaliNo ratings yet
- Daftar Pustaka Makalah ObatDocument2 pagesDaftar Pustaka Makalah Obatnur adiniNo ratings yet
- Intrathecal Antibacterial and Antifungal TherapiesDocument24 pagesIntrathecal Antibacterial and Antifungal TherapiesntnquynhproNo ratings yet
- KAPS Ques - July 2022Document2 pagesKAPS Ques - July 2022majid tariq100% (1)
- AHA Hypertension Guidelines 2017Document22 pagesAHA Hypertension Guidelines 2017Anonymous elSqPhzKNo ratings yet
- Drug Study-Operating RoomDocument6 pagesDrug Study-Operating RoomkathzheinNo ratings yet
- Malone 2013Document8 pagesMalone 2013Nguyễn LongNo ratings yet
- DRUG-STUDY PediaDocument5 pagesDRUG-STUDY PediaRhea Jane Bongcato50% (2)
- School Based Immunization Form Grade 1Document1 pageSchool Based Immunization Form Grade 1maristellaNo ratings yet
- 4 Hospital Medication OrderDocument36 pages4 Hospital Medication Orderसन्दिप क्षेत्रीNo ratings yet
- Hyoscine-N-butylbromide (HNBB)Document1 pageHyoscine-N-butylbromide (HNBB)Kyle DelrosarioNo ratings yet
- Operation FaceBOOKED ArrestsDocument6 pagesOperation FaceBOOKED ArrestsWGN Web Desk0% (1)
- Saba Hanif Presentation...Document19 pagesSaba Hanif Presentation...Saba KhanNo ratings yet
- Common Ophthalmic DrugsDocument3 pagesCommon Ophthalmic DrugsBadr DihamNo ratings yet
- DRUG STUDY 2023 WARD POTASSIUM CHLORIDE AutosavedDocument2 pagesDRUG STUDY 2023 WARD POTASSIUM CHLORIDE AutosavedMary Grace AgataNo ratings yet
- Aceclofenac Vs Diclofenac On Hexosamine LevelsDocument11 pagesAceclofenac Vs Diclofenac On Hexosamine LevelsMartin MoranNo ratings yet
- Research: Legalization of Marijuana in The PhilippinesDocument8 pagesResearch: Legalization of Marijuana in The PhilippinesPhilip Sta. AnaNo ratings yet
- Stannard Chapter5Document22 pagesStannard Chapter5Mae ann BalgoaNo ratings yet
- Print Obat Utk Ruang DokterDocument4 pagesPrint Obat Utk Ruang DokterIntan Kamala AisyiahNo ratings yet
- LECTURE NOTES For Health Science StudentDocument211 pagesLECTURE NOTES For Health Science StudentMohsinNo ratings yet
- Aripiprazole in Meige Syndrome Clinical Response.6Document4 pagesAripiprazole in Meige Syndrome Clinical Response.6tsyrahmaniNo ratings yet
- ChemSex (From A Safety Perspective)Document4 pagesChemSex (From A Safety Perspective)Dominic Milton TrottNo ratings yet
- Drug Study Ferrous Sulfate + FADocument3 pagesDrug Study Ferrous Sulfate + FAKristine ChampnessNo ratings yet
- Cefuroxime 1st PDF TRIAL PDFDocument1 pageCefuroxime 1st PDF TRIAL PDFRyanNo ratings yet
- Answer KeyDocument8 pagesAnswer KeyYap JackyNo ratings yet
- DyslipidemiaDocument11 pagesDyslipidemiaDr-Mohammed ElsawyNo ratings yet
- Pharma Drug StudyDocument56 pagesPharma Drug StudyGrace Pikit Bacsan100% (1)
- Review: Antacids in The Treatment of Gastroduodenal UlcerDocument7 pagesReview: Antacids in The Treatment of Gastroduodenal UlcerClaudia CorjăuceanuNo ratings yet
- YohimbeDocument2 pagesYohimbejssshNo ratings yet
- Montair LC TabletsDocument9 pagesMontair LC Tabletskurutala100% (1)