
Crispr Articulo XDXDXDTTTTTTTTTTTTTTTT PDF
Crispr Articulo XDXDXDTTTTTTTTTTTTTTTT PDF
You might also like
- Lab Report CRISPR Sample 1Document9 pagesLab Report CRISPR Sample 1mayagal1707No ratings yet
- Cell Engineering 101Document20 pagesCell Engineering 101Vương HoàngNo ratings yet
- Vaccination Consent Form: Tetanus, Diphtheria / Inactivated Polio Vaccine (DTP) & Meningococcal ACWY (Men ACWY)Document2 pagesVaccination Consent Form: Tetanus, Diphtheria / Inactivated Polio Vaccine (DTP) & Meningococcal ACWY (Men ACWY)meghaliNo ratings yet
- EvaluatePharma World Preview 2018Document47 pagesEvaluatePharma World Preview 2018Laura MaldonadoNo ratings yet
- Kon Erm Ann 2014Document18 pagesKon Erm Ann 2014Letícia AlibertiNo ratings yet
- CRISPR/Cas9 & Targeted Genome Editing: New Era in Molecular BiologyDocument7 pagesCRISPR/Cas9 & Targeted Genome Editing: New Era in Molecular BiologykekkaigenkeiNo ratings yet
- Crispr Cas9 and Targeted Genome EditingDocument5 pagesCrispr Cas9 and Targeted Genome EditingDaniH46No ratings yet
- BIO 254 Experiment - 2Document15 pagesBIO 254 Experiment - 2bursalibeyza494No ratings yet
- Prof Silberklang BTECH-480 Term Paper Topic-CRISPR Cas 9 Gene EditingDocument9 pagesProf Silberklang BTECH-480 Term Paper Topic-CRISPR Cas 9 Gene EditingkonarkNo ratings yet
- The FEBS Journal - 2014 - Ma - Genome Modification by CRISPR Cas9Document8 pagesThe FEBS Journal - 2014 - Ma - Genome Modification by CRISPR Cas9Alberto Luis Lizcano GonzálezNo ratings yet
- Crispr BMB402 2022Document36 pagesCrispr BMB402 2022Mohadev SahaNo ratings yet
- CRISPaint Allows Modular Base-Specific Gene TagginDocument12 pagesCRISPaint Allows Modular Base-Specific Gene TagginJyotirmayee TalapatraNo ratings yet
- Supplementary Materials For: RNA-Guided Human Genome Engineering Via Cas9Document36 pagesSupplementary Materials For: RNA-Guided Human Genome Engineering Via Cas9Ronilo Jose Danila FloresNo ratings yet
- Ma 2014Document20 pagesMa 2014hiwmacrigeeeNo ratings yet
- Addgene CRISPR GuideDocument18 pagesAddgene CRISPR GuideThomasNo ratings yet
- Genome Editing: PLNT2530 (2020) Unit 9Document25 pagesGenome Editing: PLNT2530 (2020) Unit 9Swati JainNo ratings yet
- Crispr Cas 1Document10 pagesCrispr Cas 1ABHISHEK SWARNAKARNo ratings yet
- Protein From Defluviimonas Sр.: Priority Application Title: DNA CUTTING MEANS BASED ON CAS9Document13 pagesProtein From Defluviimonas Sр.: Priority Application Title: DNA CUTTING MEANS BASED ON CAS9MichaelandKaye DanaoNo ratings yet
- Targeted Genome Editing With A DNA-dependent DNA Polymerase and Exogenous DNA-containing TemplatesDocument21 pagesTargeted Genome Editing With A DNA-dependent DNA Polymerase and Exogenous DNA-containing TemplatesSabranth GuptaNo ratings yet
- Crispr Week 12-13 by AyeshaDocument12 pagesCrispr Week 12-13 by AyeshaAyesha KhalidNo ratings yet
- Lec 2 Gene Editing Tools Part 1Document23 pagesLec 2 Gene Editing Tools Part 1mahhmoNo ratings yet
- Efficient CRISPR Editing With A Hypercompact Cas12Document14 pagesEfficient CRISPR Editing With A Hypercompact Cas12Aimen FatimaNo ratings yet
- 2021-Review-Single-Base Resolution Increasing The Specificity of The CRISPR-Cas SystemDocument12 pages2021-Review-Single-Base Resolution Increasing The Specificity of The CRISPR-Cas SystemCristian Felipe Sandoval QuiñonezNo ratings yet
- A Non-B DNA Can Replace Heptamer of V (D) J Recombination When Present Along With A Nonamer: Implications in Chromosomal Translocations and CancerDocument11 pagesA Non-B DNA Can Replace Heptamer of V (D) J Recombination When Present Along With A Nonamer: Implications in Chromosomal Translocations and CancerserioustalkNo ratings yet
- Clover KnockinDocument14 pagesClover Knockingemmen99No ratings yet
- Wolter & Putcha (2018) - Plant Transcription FactorsDocument18 pagesWolter & Putcha (2018) - Plant Transcription FactorsAna Luiza Atella de FreitasNo ratings yet
- EDICIÓN GENÉTICA CRISPRcasDocument50 pagesEDICIÓN GENÉTICA CRISPRcasMAKARENA JIMENEZ VILLEGASNo ratings yet
- Miki2021 Protocol CRISPRCas9-BasedGenomeEditingTDocument26 pagesMiki2021 Protocol CRISPRCas9-BasedGenomeEditingTSergio D. PérezNo ratings yet
- Crispr Cas9 - Google ScholarDocument1 pageCrispr Cas9 - Google ScholarPratyus GurungNo ratings yet
- CRISPR/Cas9 Platforms For Genome Editing in Plants: Developments and ApplicationsDocument14 pagesCRISPR/Cas9 Platforms For Genome Editing in Plants: Developments and ApplicationsAli HamzaNo ratings yet
- Gabr Mahmoud ThesisDocument35 pagesGabr Mahmoud Thesismahmoud gabrNo ratings yet
- Past, Present, and Future of CRISPR Genome Editing TechnologiesDocument25 pagesPast, Present, and Future of CRISPR Genome Editing TechnologiesZiyaNo ratings yet
- Nihms 1732063Document20 pagesNihms 1732063mustafa gökçenNo ratings yet
- Synthetic Guide RNA For CRISPR Genome EditingDocument9 pagesSynthetic Guide RNA For CRISPR Genome EditinggiacummoNo ratings yet
- Editing Plant Genomes With Crispr/Cas9: SciencedirectDocument9 pagesEditing Plant Genomes With Crispr/Cas9: SciencedirectVictor BonillaNo ratings yet
- N Comms 13274Document7 pagesN Comms 13274doniNo ratings yet
- (The Experimenter Series) Cornel Mulhardt - Molecular Biology and Genomics-Academic Press (2006)Document21 pages(The Experimenter Series) Cornel Mulhardt - Molecular Biology and Genomics-Academic Press (2006)Teflon SlimNo ratings yet
- Science Aba8853Document8 pagesScience Aba8853ירדן לויןNo ratings yet
- Advances in CRISPR-Cas9 Genome Engineering: Lessons Learned From RNA InterferenceDocument13 pagesAdvances in CRISPR-Cas9 Genome Engineering: Lessons Learned From RNA InterferenceAmina Tucak-SmajićNo ratings yet
- Park Et Al. 2014Document8 pagesPark Et Al. 2014Benedikt EngelNo ratings yet
- ZFN, Talens, Crispr PDFDocument9 pagesZFN, Talens, Crispr PDFjezelle lividNo ratings yet
- 2019 - Controlling CRISPR-Cas9 With Ligand-ActivatedDocument11 pages2019 - Controlling CRISPR-Cas9 With Ligand-ActivatedFrani Villagran SilvaNo ratings yet
- Cas 9Document28 pagesCas 9Rin ChanNo ratings yet
- Anders Et Al., 2014Document22 pagesAnders Et Al., 2014Tatiana Sanchez AlvarezNo ratings yet
- Genome EditingDocument12 pagesGenome EditingDahlia StudioNo ratings yet
- CRISPR/Cas9 For Genome Editing: Progress, Implications and ChallengesDocument7 pagesCRISPR/Cas9 For Genome Editing: Progress, Implications and ChallengesAngelica RestrepoNo ratings yet
- RNA Sequencing: An Introduction To Efficient Planning and Execution of RNA Sequencing (RNA-Seq) ExperimentsDocument6 pagesRNA Sequencing: An Introduction To Efficient Planning and Execution of RNA Sequencing (RNA-Seq) ExperimentsnareshNo ratings yet
- Infographic - CRISPR - Cas9Document3 pagesInfographic - CRISPR - Cas9felipe duarteNo ratings yet
- Method and Result:: 2.1. Sg-RNA DesigningDocument9 pagesMethod and Result:: 2.1. Sg-RNA DesigningMasum Billah TuhinNo ratings yet
- CRISPR/Cas9 Mediated Targeting of Multiple Genes in DictyosteliumDocument11 pagesCRISPR/Cas9 Mediated Targeting of Multiple Genes in DictyosteliumIram AmmadNo ratings yet
- Grainger 2016Document4 pagesGrainger 2016SergiNo ratings yet
- A Protocol For Designing Sirnas With High Functionality and SpecificityDocument11 pagesA Protocol For Designing Sirnas With High Functionality and SpecificityThoha AlbaarNo ratings yet
- ReportDocument6 pagesReportramna ziaNo ratings yet
- CRISPR EbookDocument14 pagesCRISPR EbookBayan MallahNo ratings yet
- Crispr Jove PDFDocument10 pagesCrispr Jove PDFRupendra ShresthaNo ratings yet
- Crispr 101: Your Guide To Understanding CRISPRDocument29 pagesCrispr 101: Your Guide To Understanding CRISPRydydNo ratings yet
- Vakulskas Behlke 2019 Evaluation and Reduction of Crispr Off Target Cleavage EventsDocument8 pagesVakulskas Behlke 2019 Evaluation and Reduction of Crispr Off Target Cleavage Eventsירדן לויןNo ratings yet
- Genome Editing and Crispr Cas 9Document12 pagesGenome Editing and Crispr Cas 9HemsaiYadavNo ratings yet
- A Family of Lambda Phage cDNA Cloning VectorsDocument10 pagesA Family of Lambda Phage cDNA Cloning VectorsaaasidNo ratings yet
- 1 gRNADocument6 pages1 gRNA冯博士No ratings yet
- Gene Editing Prime Time 10.1056@NEJMcibr1914271Document4 pagesGene Editing Prime Time 10.1056@NEJMcibr1914271MCuk2606No ratings yet
- The CRISPR-Cas9 Toolbox: Applications and Ethical Considerations.From EverandThe CRISPR-Cas9 Toolbox: Applications and Ethical Considerations.No ratings yet
- Molecular Biology of The Cell Sixth EditionDocument3 pagesMolecular Biology of The Cell Sixth EditionygutierrezmNo ratings yet
- Guide To Nucleic Acid Purification-2Document6 pagesGuide To Nucleic Acid Purification-2DolphingNo ratings yet
- Lab Topic 14 QA & QC in The Molecular LaboratoryDocument6 pagesLab Topic 14 QA & QC in The Molecular LaboratoryNatasha MendozaNo ratings yet
- Active TransportDocument8 pagesActive Transportapi-344568500No ratings yet
- Biochemistry Lec 1Document43 pagesBiochemistry Lec 1ao868598No ratings yet
- 12th Bio Botany EM Important Questions English Medium PDF DownloadDocument7 pages12th Bio Botany EM Important Questions English Medium PDF DownloadPrase 1815No ratings yet
- Applications of Biotechnology in The IndustryDocument6 pagesApplications of Biotechnology in The IndustryAlezandraNo ratings yet
- Physiology AssignmentDocument8 pagesPhysiology Assignmentmalokkhan183No ratings yet
- Ecosystem: Life Energy: PhotosynthesisDocument8 pagesEcosystem: Life Energy: PhotosynthesisHelma Jabello AriolaNo ratings yet
- Genetic Diversity and Population Genetic Analysis of Donax Vittatus and Phylogeny of The Genus With Mitocondrial and Nuclear MarkersDocument10 pagesGenetic Diversity and Population Genetic Analysis of Donax Vittatus and Phylogeny of The Genus With Mitocondrial and Nuclear MarkersMarco Antonio SolisNo ratings yet
- Bio 214 Syllabus - Fall 2020Document27 pagesBio 214 Syllabus - Fall 2020Kate Robb AllmanNo ratings yet
- PCR Guided Notes AfterDocument2 pagesPCR Guided Notes Afterjbhuffman75No ratings yet
- Genetic Circuit Automation - Alec NielsenDocument13 pagesGenetic Circuit Automation - Alec NielsenPablo Cardona PerezNo ratings yet
- 1 10 2020 Transcription and Translation - KnexDocument4 pages1 10 2020 Transcription and Translation - Knexapi-508716694No ratings yet
- User Guide: Targetron Gene Knockout SystemDocument28 pagesUser Guide: Targetron Gene Knockout SystemLuisNo ratings yet
- DNA ReplicationDocument13 pagesDNA ReplicationJoanne NgauNo ratings yet
- First Generation SequencingDocument20 pagesFirst Generation SequencingWinrad D. Velarde100% (1)
- Lesson Title: B13.4 DNA and The Human Genome Project: Connector: Questions On Prior Learning Linked To Todays LessonDocument46 pagesLesson Title: B13.4 DNA and The Human Genome Project: Connector: Questions On Prior Learning Linked To Todays LessonBenjamin WatsonNo ratings yet
- Senate Hearing, 109TH Congress - Strengthening Participation of Small Businesses in Federal Contracting and Innovation Research ProgramsDocument238 pagesSenate Hearing, 109TH Congress - Strengthening Participation of Small Businesses in Federal Contracting and Innovation Research ProgramsScribd Government DocsNo ratings yet
- Research Proposal FinalDocument6 pagesResearch Proposal Finalapi-311572504No ratings yet
- Longevity ReportDocument3 pagesLongevity ReportAlvatrox MarcNo ratings yet
- September 26, 2018 by Sagar Aryal: Antigen-Properties, Types and Determinants of AntigenicityDocument23 pagesSeptember 26, 2018 by Sagar Aryal: Antigen-Properties, Types and Determinants of AntigenicityEzekiel GantiedNo ratings yet
- M.SC Ag Fruit ScienceDocument21 pagesM.SC Ag Fruit ScienceBhuvanaNo ratings yet
- What Is A Chemical EngineerDocument4 pagesWhat Is A Chemical EngineerJose Antonio Fermin JimenezNo ratings yet
- Biology Module 5 NotesDocument17 pagesBiology Module 5 NotesKatie BellNo ratings yet
- PAIIadvKulkarniSengupta (NXPowerLite Copy)Document2 pagesPAIIadvKulkarniSengupta (NXPowerLite Copy)Baba KumarNo ratings yet
- SAC Review: DNA Methylation: A Form of Epigenetic Control of Gene ExpressionDocument6 pagesSAC Review: DNA Methylation: A Form of Epigenetic Control of Gene ExpressionchumalakaNo ratings yet
- Brain Energy RescueDocument25 pagesBrain Energy RescueMaria Vitória Cota de AbreuNo ratings yet










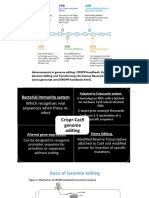



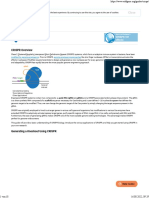











































































Download as docx, pdf, or txt
You might also like
- Lab Report CRISPR Sample 1Document9 pagesLab Report CRISPR Sample 1mayagal1707No ratings yet
- Cell Engineering 101Document20 pagesCell Engineering 101Vương HoàngNo ratings yet
- Vaccination Consent Form: Tetanus, Diphtheria / Inactivated Polio Vaccine (DTP) & Meningococcal ACWY (Men ACWY)Document2 pagesVaccination Consent Form: Tetanus, Diphtheria / Inactivated Polio Vaccine (DTP) & Meningococcal ACWY (Men ACWY)meghaliNo ratings yet
- EvaluatePharma World Preview 2018Document47 pagesEvaluatePharma World Preview 2018Laura MaldonadoNo ratings yet
- Kon Erm Ann 2014Document18 pagesKon Erm Ann 2014Letícia AlibertiNo ratings yet
- CRISPR/Cas9 & Targeted Genome Editing: New Era in Molecular BiologyDocument7 pagesCRISPR/Cas9 & Targeted Genome Editing: New Era in Molecular BiologykekkaigenkeiNo ratings yet
- Crispr Cas9 and Targeted Genome EditingDocument5 pagesCrispr Cas9 and Targeted Genome EditingDaniH46No ratings yet
- BIO 254 Experiment - 2Document15 pagesBIO 254 Experiment - 2bursalibeyza494No ratings yet
- Prof Silberklang BTECH-480 Term Paper Topic-CRISPR Cas 9 Gene EditingDocument9 pagesProf Silberklang BTECH-480 Term Paper Topic-CRISPR Cas 9 Gene EditingkonarkNo ratings yet
- The FEBS Journal - 2014 - Ma - Genome Modification by CRISPR Cas9Document8 pagesThe FEBS Journal - 2014 - Ma - Genome Modification by CRISPR Cas9Alberto Luis Lizcano GonzálezNo ratings yet
- Crispr BMB402 2022Document36 pagesCrispr BMB402 2022Mohadev SahaNo ratings yet
- CRISPaint Allows Modular Base-Specific Gene TagginDocument12 pagesCRISPaint Allows Modular Base-Specific Gene TagginJyotirmayee TalapatraNo ratings yet
- Supplementary Materials For: RNA-Guided Human Genome Engineering Via Cas9Document36 pagesSupplementary Materials For: RNA-Guided Human Genome Engineering Via Cas9Ronilo Jose Danila FloresNo ratings yet
- Ma 2014Document20 pagesMa 2014hiwmacrigeeeNo ratings yet
- Addgene CRISPR GuideDocument18 pagesAddgene CRISPR GuideThomasNo ratings yet
- Genome Editing: PLNT2530 (2020) Unit 9Document25 pagesGenome Editing: PLNT2530 (2020) Unit 9Swati JainNo ratings yet
- Crispr Cas 1Document10 pagesCrispr Cas 1ABHISHEK SWARNAKARNo ratings yet
- Protein From Defluviimonas Sр.: Priority Application Title: DNA CUTTING MEANS BASED ON CAS9Document13 pagesProtein From Defluviimonas Sр.: Priority Application Title: DNA CUTTING MEANS BASED ON CAS9MichaelandKaye DanaoNo ratings yet
- Targeted Genome Editing With A DNA-dependent DNA Polymerase and Exogenous DNA-containing TemplatesDocument21 pagesTargeted Genome Editing With A DNA-dependent DNA Polymerase and Exogenous DNA-containing TemplatesSabranth GuptaNo ratings yet
- Crispr Week 12-13 by AyeshaDocument12 pagesCrispr Week 12-13 by AyeshaAyesha KhalidNo ratings yet
- Lec 2 Gene Editing Tools Part 1Document23 pagesLec 2 Gene Editing Tools Part 1mahhmoNo ratings yet
- Efficient CRISPR Editing With A Hypercompact Cas12Document14 pagesEfficient CRISPR Editing With A Hypercompact Cas12Aimen FatimaNo ratings yet
- 2021-Review-Single-Base Resolution Increasing The Specificity of The CRISPR-Cas SystemDocument12 pages2021-Review-Single-Base Resolution Increasing The Specificity of The CRISPR-Cas SystemCristian Felipe Sandoval QuiñonezNo ratings yet
- A Non-B DNA Can Replace Heptamer of V (D) J Recombination When Present Along With A Nonamer: Implications in Chromosomal Translocations and CancerDocument11 pagesA Non-B DNA Can Replace Heptamer of V (D) J Recombination When Present Along With A Nonamer: Implications in Chromosomal Translocations and CancerserioustalkNo ratings yet
- Clover KnockinDocument14 pagesClover Knockingemmen99No ratings yet
- Wolter & Putcha (2018) - Plant Transcription FactorsDocument18 pagesWolter & Putcha (2018) - Plant Transcription FactorsAna Luiza Atella de FreitasNo ratings yet
- EDICIÓN GENÉTICA CRISPRcasDocument50 pagesEDICIÓN GENÉTICA CRISPRcasMAKARENA JIMENEZ VILLEGASNo ratings yet
- Miki2021 Protocol CRISPRCas9-BasedGenomeEditingTDocument26 pagesMiki2021 Protocol CRISPRCas9-BasedGenomeEditingTSergio D. PérezNo ratings yet
- Crispr Cas9 - Google ScholarDocument1 pageCrispr Cas9 - Google ScholarPratyus GurungNo ratings yet
- CRISPR/Cas9 Platforms For Genome Editing in Plants: Developments and ApplicationsDocument14 pagesCRISPR/Cas9 Platforms For Genome Editing in Plants: Developments and ApplicationsAli HamzaNo ratings yet
- Gabr Mahmoud ThesisDocument35 pagesGabr Mahmoud Thesismahmoud gabrNo ratings yet
- Past, Present, and Future of CRISPR Genome Editing TechnologiesDocument25 pagesPast, Present, and Future of CRISPR Genome Editing TechnologiesZiyaNo ratings yet
- Nihms 1732063Document20 pagesNihms 1732063mustafa gökçenNo ratings yet
- Synthetic Guide RNA For CRISPR Genome EditingDocument9 pagesSynthetic Guide RNA For CRISPR Genome EditinggiacummoNo ratings yet
- Editing Plant Genomes With Crispr/Cas9: SciencedirectDocument9 pagesEditing Plant Genomes With Crispr/Cas9: SciencedirectVictor BonillaNo ratings yet
- N Comms 13274Document7 pagesN Comms 13274doniNo ratings yet
- (The Experimenter Series) Cornel Mulhardt - Molecular Biology and Genomics-Academic Press (2006)Document21 pages(The Experimenter Series) Cornel Mulhardt - Molecular Biology and Genomics-Academic Press (2006)Teflon SlimNo ratings yet
- Science Aba8853Document8 pagesScience Aba8853ירדן לויןNo ratings yet
- Advances in CRISPR-Cas9 Genome Engineering: Lessons Learned From RNA InterferenceDocument13 pagesAdvances in CRISPR-Cas9 Genome Engineering: Lessons Learned From RNA InterferenceAmina Tucak-SmajićNo ratings yet
- Park Et Al. 2014Document8 pagesPark Et Al. 2014Benedikt EngelNo ratings yet
- ZFN, Talens, Crispr PDFDocument9 pagesZFN, Talens, Crispr PDFjezelle lividNo ratings yet
- 2019 - Controlling CRISPR-Cas9 With Ligand-ActivatedDocument11 pages2019 - Controlling CRISPR-Cas9 With Ligand-ActivatedFrani Villagran SilvaNo ratings yet
- Cas 9Document28 pagesCas 9Rin ChanNo ratings yet
- Anders Et Al., 2014Document22 pagesAnders Et Al., 2014Tatiana Sanchez AlvarezNo ratings yet
- Genome EditingDocument12 pagesGenome EditingDahlia StudioNo ratings yet
- CRISPR/Cas9 For Genome Editing: Progress, Implications and ChallengesDocument7 pagesCRISPR/Cas9 For Genome Editing: Progress, Implications and ChallengesAngelica RestrepoNo ratings yet
- RNA Sequencing: An Introduction To Efficient Planning and Execution of RNA Sequencing (RNA-Seq) ExperimentsDocument6 pagesRNA Sequencing: An Introduction To Efficient Planning and Execution of RNA Sequencing (RNA-Seq) ExperimentsnareshNo ratings yet
- Infographic - CRISPR - Cas9Document3 pagesInfographic - CRISPR - Cas9felipe duarteNo ratings yet
- Method and Result:: 2.1. Sg-RNA DesigningDocument9 pagesMethod and Result:: 2.1. Sg-RNA DesigningMasum Billah TuhinNo ratings yet
- CRISPR/Cas9 Mediated Targeting of Multiple Genes in DictyosteliumDocument11 pagesCRISPR/Cas9 Mediated Targeting of Multiple Genes in DictyosteliumIram AmmadNo ratings yet
- Grainger 2016Document4 pagesGrainger 2016SergiNo ratings yet
- A Protocol For Designing Sirnas With High Functionality and SpecificityDocument11 pagesA Protocol For Designing Sirnas With High Functionality and SpecificityThoha AlbaarNo ratings yet
- ReportDocument6 pagesReportramna ziaNo ratings yet
- CRISPR EbookDocument14 pagesCRISPR EbookBayan MallahNo ratings yet
- Crispr Jove PDFDocument10 pagesCrispr Jove PDFRupendra ShresthaNo ratings yet
- Crispr 101: Your Guide To Understanding CRISPRDocument29 pagesCrispr 101: Your Guide To Understanding CRISPRydydNo ratings yet
- Vakulskas Behlke 2019 Evaluation and Reduction of Crispr Off Target Cleavage EventsDocument8 pagesVakulskas Behlke 2019 Evaluation and Reduction of Crispr Off Target Cleavage Eventsירדן לויןNo ratings yet
- Genome Editing and Crispr Cas 9Document12 pagesGenome Editing and Crispr Cas 9HemsaiYadavNo ratings yet
- A Family of Lambda Phage cDNA Cloning VectorsDocument10 pagesA Family of Lambda Phage cDNA Cloning VectorsaaasidNo ratings yet
- 1 gRNADocument6 pages1 gRNA冯博士No ratings yet
- Gene Editing Prime Time 10.1056@NEJMcibr1914271Document4 pagesGene Editing Prime Time 10.1056@NEJMcibr1914271MCuk2606No ratings yet
- The CRISPR-Cas9 Toolbox: Applications and Ethical Considerations.From EverandThe CRISPR-Cas9 Toolbox: Applications and Ethical Considerations.No ratings yet
- Molecular Biology of The Cell Sixth EditionDocument3 pagesMolecular Biology of The Cell Sixth EditionygutierrezmNo ratings yet
- Guide To Nucleic Acid Purification-2Document6 pagesGuide To Nucleic Acid Purification-2DolphingNo ratings yet
- Lab Topic 14 QA & QC in The Molecular LaboratoryDocument6 pagesLab Topic 14 QA & QC in The Molecular LaboratoryNatasha MendozaNo ratings yet
- Active TransportDocument8 pagesActive Transportapi-344568500No ratings yet
- Biochemistry Lec 1Document43 pagesBiochemistry Lec 1ao868598No ratings yet
- 12th Bio Botany EM Important Questions English Medium PDF DownloadDocument7 pages12th Bio Botany EM Important Questions English Medium PDF DownloadPrase 1815No ratings yet
- Applications of Biotechnology in The IndustryDocument6 pagesApplications of Biotechnology in The IndustryAlezandraNo ratings yet
- Physiology AssignmentDocument8 pagesPhysiology Assignmentmalokkhan183No ratings yet
- Ecosystem: Life Energy: PhotosynthesisDocument8 pagesEcosystem: Life Energy: PhotosynthesisHelma Jabello AriolaNo ratings yet
- Genetic Diversity and Population Genetic Analysis of Donax Vittatus and Phylogeny of The Genus With Mitocondrial and Nuclear MarkersDocument10 pagesGenetic Diversity and Population Genetic Analysis of Donax Vittatus and Phylogeny of The Genus With Mitocondrial and Nuclear MarkersMarco Antonio SolisNo ratings yet
- Bio 214 Syllabus - Fall 2020Document27 pagesBio 214 Syllabus - Fall 2020Kate Robb AllmanNo ratings yet
- PCR Guided Notes AfterDocument2 pagesPCR Guided Notes Afterjbhuffman75No ratings yet
- Genetic Circuit Automation - Alec NielsenDocument13 pagesGenetic Circuit Automation - Alec NielsenPablo Cardona PerezNo ratings yet
- 1 10 2020 Transcription and Translation - KnexDocument4 pages1 10 2020 Transcription and Translation - Knexapi-508716694No ratings yet
- User Guide: Targetron Gene Knockout SystemDocument28 pagesUser Guide: Targetron Gene Knockout SystemLuisNo ratings yet
- DNA ReplicationDocument13 pagesDNA ReplicationJoanne NgauNo ratings yet
- First Generation SequencingDocument20 pagesFirst Generation SequencingWinrad D. Velarde100% (1)
- Lesson Title: B13.4 DNA and The Human Genome Project: Connector: Questions On Prior Learning Linked To Todays LessonDocument46 pagesLesson Title: B13.4 DNA and The Human Genome Project: Connector: Questions On Prior Learning Linked To Todays LessonBenjamin WatsonNo ratings yet
- Senate Hearing, 109TH Congress - Strengthening Participation of Small Businesses in Federal Contracting and Innovation Research ProgramsDocument238 pagesSenate Hearing, 109TH Congress - Strengthening Participation of Small Businesses in Federal Contracting and Innovation Research ProgramsScribd Government DocsNo ratings yet
- Research Proposal FinalDocument6 pagesResearch Proposal Finalapi-311572504No ratings yet
- Longevity ReportDocument3 pagesLongevity ReportAlvatrox MarcNo ratings yet
- September 26, 2018 by Sagar Aryal: Antigen-Properties, Types and Determinants of AntigenicityDocument23 pagesSeptember 26, 2018 by Sagar Aryal: Antigen-Properties, Types and Determinants of AntigenicityEzekiel GantiedNo ratings yet
- M.SC Ag Fruit ScienceDocument21 pagesM.SC Ag Fruit ScienceBhuvanaNo ratings yet
- What Is A Chemical EngineerDocument4 pagesWhat Is A Chemical EngineerJose Antonio Fermin JimenezNo ratings yet
- Biology Module 5 NotesDocument17 pagesBiology Module 5 NotesKatie BellNo ratings yet
- PAIIadvKulkarniSengupta (NXPowerLite Copy)Document2 pagesPAIIadvKulkarniSengupta (NXPowerLite Copy)Baba KumarNo ratings yet
- SAC Review: DNA Methylation: A Form of Epigenetic Control of Gene ExpressionDocument6 pagesSAC Review: DNA Methylation: A Form of Epigenetic Control of Gene ExpressionchumalakaNo ratings yet
- Brain Energy RescueDocument25 pagesBrain Energy RescueMaria Vitória Cota de AbreuNo ratings yet