Download as pdf or txt
You might also like
- US Contempt Judgment On Bongbong Marcos RedactedDocument14 pagesUS Contempt Judgment On Bongbong Marcos RedactedRappler100% (1)
- Quadri Et Al. PROCESS FOR THE PRODUCTION OF EPDM ELASTOMERS IN SOLUTION AND POLYMERISATION REACTORDocument32 pagesQuadri Et Al. PROCESS FOR THE PRODUCTION OF EPDM ELASTOMERS IN SOLUTION AND POLYMERISATION REACTORJohn Patrick DagleNo ratings yet
- Essay ExploratoryDocument6 pagesEssay Exploratoryapi-240678482No ratings yet
- World: International BureauDocument15 pagesWorld: International BureauAdhisya Salma KhairunnisaNo ratings yet
- WO2007069266A2Document13 pagesWO2007069266A2Dr. Narsinh DodiyaNo ratings yet
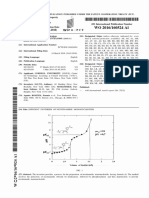
- WO 2013/087238 Al: International BureauDocument24 pagesWO 2013/087238 Al: International Bureaujulianpellegrini860No ratings yet
- Wo2014195249a1 PDFDocument15 pagesWo2014195249a1 PDFAnonymous DxaBg8fUENo ratings yet
- Process For The Polymerization of Vinyl ChlorideDocument32 pagesProcess For The Polymerization of Vinyl ChlorideThu TongNo ratings yet
- International BureauDocument13 pagesInternational Bureautsilavo solofonomenaNo ratings yet
- International Bureau: (19) World Intellectual Property OrganizationDocument22 pagesInternational Bureau: (19) World Intellectual Property OrganizationVana BamNo ratings yet
- WO2016057639A1Document36 pagesWO2016057639A1هیمن مNo ratings yet
- NaOH AlkalinasiDocument30 pagesNaOH AlkalinasiYonatan AdiNo ratings yet
- WO2015189857A1Document31 pagesWO2015189857A1Nguyễn Xuân TùngNo ratings yet
- Ciprofloxacin PDFDocument20 pagesCiprofloxacin PDFnurvaniNo ratings yet
- WO2009004593A2Document13 pagesWO2009004593A2researchNo ratings yet
- WO2016102499A1Document71 pagesWO2016102499A1梅汉No ratings yet
- 20 September 2007 (20.09.2007) : (19) World Intellectual Property OrganizationDocument20 pages20 September 2007 (20.09.2007) : (19) World Intellectual Property OrganizationCarlos SantiagoNo ratings yet
- Waste Rubber Composition - WO2021152109 - A1Document18 pagesWaste Rubber Composition - WO2021152109 - A1karim.rashidmaneshNo ratings yet
- WO2014188453A2Document43 pagesWO2014188453A2Dr-Nilesh SalunkheNo ratings yet
- QuinineDihydrochlorid DirectExtractionDocument12 pagesQuinineDihydrochlorid DirectExtractionJozsef GaborNo ratings yet
- 2.5 PH Water Disinfectant PDFDocument15 pages2.5 PH Water Disinfectant PDFEry KaNo ratings yet
- WO2017121980A1Document24 pagesWO2017121980A1Hardimas dwi cahyoNo ratings yet
- N Hexyl LiteraturDocument40 pagesN Hexyl LiteraturVivian DoNo ratings yet
- WO2009153654A1Document22 pagesWO2009153654A1jihanvrpNo ratings yet
- WO2011151724A2Document45 pagesWO2011151724A2ungureanucameliaNo ratings yet
- Cele CoxDocument15 pagesCele CoxLinsay Granados CondeNo ratings yet
- Standard Patent (11) Application No. AU 2010283544 B2 (19) Australian Patent OfficeDocument12 pagesStandard Patent (11) Application No. AU 2010283544 B2 (19) Australian Patent OfficeChoo Wei shengNo ratings yet
- Cobalto PatenteDocument26 pagesCobalto PatentemiriamNo ratings yet
- International BureauDocument28 pagesInternational BureauNace AtanasovNo ratings yet
- World: (19) Intellectual Property Organization (10) International Publication Number (43) International Publication DateDocument21 pagesWorld: (19) Intellectual Property Organization (10) International Publication Number (43) International Publication DateshaNo ratings yet
- Patent Hand SanitizerDocument15 pagesPatent Hand SanitizerFebrianto DimazNo ratings yet
- WO2013049605A1Document33 pagesWO2013049605A1nagarajharishNo ratings yet
- International BureauDocument25 pagesInternational BureauKaboy PumNo ratings yet
- D 3Document23 pagesD 3Anonymous C3BD7OdNo ratings yet
- International BureauDocument24 pagesInternational BureauniteshacharyaNo ratings yet
- View PDFDocument29 pagesView PDFADARSH PNo ratings yet
- International BureauDocument33 pagesInternational BureauBobNo ratings yet
- Spray Dried FlavourDocument22 pagesSpray Dried FlavourAlex HahnNo ratings yet
- WO2010069028A1Document26 pagesWO2010069028A1NnaLupizNo ratings yet
- WO2018075145A1Document49 pagesWO2018075145A1samik4uNo ratings yet
- World Intellectual Property Organization International Publication Number International Publication Date 17 February 2011 (17.02.2011)Document30 pagesWorld Intellectual Property Organization International Publication Number International Publication Date 17 February 2011 (17.02.2011)mailbabuNo ratings yet
- Au2016354228b2 Seed CoatingDocument28 pagesAu2016354228b2 Seed CoatingMelisa MonerrisNo ratings yet
- Wipoipct: International BureauDocument20 pagesWipoipct: International BureaujoseNo ratings yet
- Wo 2014185872 A 1Document11 pagesWo 2014185872 A 1Shahid AliNo ratings yet
- Patent-Composition Comprising Lactic Acid Bacteria For Use in The Preventive and or Curative Treatment of Bacterial VaginosisDocument28 pagesPatent-Composition Comprising Lactic Acid Bacteria For Use in The Preventive and or Curative Treatment of Bacterial VaginosisLucas LE-DOANNo ratings yet
- Epoxy Reactive DiluentsDocument27 pagesEpoxy Reactive DiluentsAlejandro Sánchez MartínezNo ratings yet
- WO2017136634A1Document24 pagesWO2017136634A1Ilyas FaizNo ratings yet
- WO2013093093A1Document17 pagesWO2013093093A1Lwanga Um NdongoNo ratings yet
- WO 2009/109985 Al: September 2009 (11.09.2009)Document25 pagesWO 2009/109985 Al: September 2009 (11.09.2009)Ornevalde JulphinNo ratings yet
- Paten 1Document23 pagesPaten 1Bagja Malik SyakurNo ratings yet
- WO 2008/074667 Al: International BureauDocument23 pagesWO 2008/074667 Al: International BureauManuel MontañoNo ratings yet
- International BureauDocument44 pagesInternational BureauRibautNo ratings yet
- Patent 1 - WO2017122216A1Document61 pagesPatent 1 - WO2017122216A1sci pusatNo ratings yet
- WO 2021/158698 Al: (10) International Publication NumberDocument234 pagesWO 2021/158698 Al: (10) International Publication Numberyoganayagi209No ratings yet
- International Application Status ReportDocument2 pagesInternational Application Status ReportshamimNo ratings yet
- Topical Formulations of HeparinDocument29 pagesTopical Formulations of HeparinAyuNo ratings yet
- WO2017122020A1Document47 pagesWO2017122020A1Nuzhat CollectionsNo ratings yet
- As To To Be 4.1 As To The To The The (Rule 4.1 (In) ) (Rule 7 (Iv) )Document11 pagesAs To To Be 4.1 As To The To The The (Rule 4.1 (In) ) (Rule 7 (Iv) )Ujang MinangNo ratings yet
- Wo2012151438a1 PDFDocument29 pagesWo2012151438a1 PDFsasibhushanarao poolaNo ratings yet
- Ep2629690 A2Document104 pagesEp2629690 A2Anton TsyrulnykovNo ratings yet
- Lubricante ConcentradoDocument20 pagesLubricante ConcentradoMónica AlboresNo ratings yet
- Packaging for Nonthermal Processing of FoodFrom EverandPackaging for Nonthermal Processing of FoodMelvin A. PascallNo ratings yet
- Permix Catalog Revised 220417 LRDocument78 pagesPermix Catalog Revised 220417 LRCory AmeliaNo ratings yet
- Fatty Acids: Avid NnekenDocument44 pagesFatty Acids: Avid NnekenCory AmeliaNo ratings yet
- CN103880632A Calcium Stearate Preparation TechnologyDocument3 pagesCN103880632A Calcium Stearate Preparation TechnologyCory AmeliaNo ratings yet
- CN103276625A Method For Preparing Calcium Stearate Water-Based Dispersion LiquidDocument5 pagesCN103276625A Method For Preparing Calcium Stearate Water-Based Dispersion LiquidCory AmeliaNo ratings yet
- Ziosi 2014Document10 pagesZiosi 2014Cory AmeliaNo ratings yet
- 2-Chloroethanol MSDSDocument8 pages2-Chloroethanol MSDSCory AmeliaNo ratings yet
- Synthesis of Aluminium TrihydrateDocument3 pagesSynthesis of Aluminium TrihydrateCory Amelia100% (1)
- CN102875356A - Calcium Stearate Production Method - Google PatentsDocument6 pagesCN102875356A - Calcium Stearate Production Method - Google PatentsCory AmeliaNo ratings yet
- Strand (11) MBDA - Subcontracts To Ministry of Defense - Manufactures WeaponsDocument3 pagesStrand (11) MBDA - Subcontracts To Ministry of Defense - Manufactures WeaponsJohn Adam St Gang: Crown Control100% (1)
- Nashville Police Complains About Secret ServiceDocument8 pagesNashville Police Complains About Secret ServiceJeffrey DunetzNo ratings yet
- Naming A ChildDocument2 pagesNaming A ChildTAQWA (Singapore)100% (5)
- In The City Civil Court, at Agra Civil Suit No. 100 of 2002Document3 pagesIn The City Civil Court, at Agra Civil Suit No. 100 of 2002Ahmed ShujaNo ratings yet
- Annexure 1 Guidelines For Loan Candidates WBODocument7 pagesAnnexure 1 Guidelines For Loan Candidates WBOwanjariabhi6235No ratings yet
- RPH AdditionalDocument58 pagesRPH AdditionalMirard A. MagsipocNo ratings yet
- Urban Informal SectorDocument14 pagesUrban Informal SectorArnel MangilimanNo ratings yet
- FM-CSVlrd-08 CPECs Joint Affidavit Rev 2 January 01 2022 25jan2022Document1 pageFM-CSVlrd-08 CPECs Joint Affidavit Rev 2 January 01 2022 25jan2022Charlene Joyce PataludNo ratings yet
- Main DefendantDocument19 pagesMain DefendantAshutosh KumarNo ratings yet
- Myron Abbott An Enduring Legagy Immigrant PioneersDocument8 pagesMyron Abbott An Enduring Legagy Immigrant PioneersshawfamilyhistoryNo ratings yet
- Food Safety AppealDocument23 pagesFood Safety Appealabhijeet panchariyaNo ratings yet
- Chapter 12 Solutions ManualDocument68 pagesChapter 12 Solutions Manualmnarv880% (1)
- Business LawDocument47 pagesBusiness LawLinh VũNo ratings yet
- Asset Based Finance Project ReportDocument52 pagesAsset Based Finance Project ReportkamdicaNo ratings yet
- Sotto v. Sotto, GR L-17768, Sept. 1, 1922, 43 Phil. 688Document1 pageSotto v. Sotto, GR L-17768, Sept. 1, 1922, 43 Phil. 688Gia Dimayuga100% (1)
- Ballast Water Convention Edition 2004 PDFDocument154 pagesBallast Water Convention Edition 2004 PDFAlfredoLopezNo ratings yet
- Evaluation Jcertificative - TESS - 2012 - Histoire - Questionnaires Et Portefeuille de Documents (Ressource 9225)Document21 pagesEvaluation Jcertificative - TESS - 2012 - Histoire - Questionnaires Et Portefeuille de Documents (Ressource 9225)joelelodieNo ratings yet
- Sanatana Dharma: Kamokarsheet Japam and Seshadri SwamigalDocument2 pagesSanatana Dharma: Kamokarsheet Japam and Seshadri SwamigalBalaji JayaramanNo ratings yet
- Argicutural Lien PDFDocument36 pagesArgicutural Lien PDFlifeisgrand100% (3)
- Confused Flour Beetle Andamp Red Flour Beetle Sds 2Document6 pagesConfused Flour Beetle Andamp Red Flour Beetle Sds 2Aardwolf PestkareNo ratings yet
- Spreading of Western Education During British RuleDocument4 pagesSpreading of Western Education During British RuleIshfaque AhmedNo ratings yet
- Assignment No.3 - Case DigestsDocument28 pagesAssignment No.3 - Case DigestsaldehNo ratings yet
- Service Bye LawsDocument94 pagesService Bye Lawssahilbansal103No ratings yet
- 1 - Baranda V Gustilo - Julius GuzmanDocument2 pages1 - Baranda V Gustilo - Julius GuzmanJulius ManaloNo ratings yet
- BookDocument5 pagesBookGodfrey Loth Sales Alcansare Jr.No ratings yet
- Birth Statistics, Philippines 2018Document182 pagesBirth Statistics, Philippines 2018GwynethAdrienneLeighVillapandoNo ratings yet
- LET Reviewer Gen. EdDocument6 pagesLET Reviewer Gen. Edsheena san gabrielNo ratings yet
- IRS Seminar Level 3, Form #12.034Document452 pagesIRS Seminar Level 3, Form #12.034Sovereignty Education and Defense Ministry (SEDM)No ratings yet