Download as pdf or txt
You might also like
- Runge KuttaDocument3 pagesRunge KuttaAllen Jerry Aries100% (1)
- Lecture NotesDocument80 pagesLecture Notes8ienneNo ratings yet
- Exp 9 GRP 9 20110153Document12 pagesExp 9 GRP 9 20110153Rahul KumarNo ratings yet
- Experiment 3: The Enthalpy of Reaction For The Dissolution of SaltsDocument12 pagesExperiment 3: The Enthalpy of Reaction For The Dissolution of Saltsleo besaNo ratings yet
- ThermochemistryDocument8 pagesThermochemistryAbdul BokhariNo ratings yet
- 111 13EnthalpyHydrationSummer2019Document7 pages111 13EnthalpyHydrationSummer2019Marco MorelliNo ratings yet
- Phase EquilibriaDocument21 pagesPhase EquilibriasuperchellyNo ratings yet
- Lab. Vapor PressureDocument4 pagesLab. Vapor PressureYiğit IlgazNo ratings yet
- Notes On Q M C Delta TDocument2 pagesNotes On Q M C Delta TDing30180No ratings yet
- Fluid and Thermal ExitDocument25 pagesFluid and Thermal Exitdavididosa40No ratings yet
- Lab ManualsDocument33 pagesLab ManualsHoai Anh NgoNo ratings yet
- Solucionario KoretsskyDocument130 pagesSolucionario KoretsskyWilliam Camilo Díaz GonzálezNo ratings yet
- TP Exp 3Document7 pagesTP Exp 3MahainiIm RuzailyNo ratings yet
- Energies: An Optimum Enthalpy Approach For Melting and Solidification With Volume ChangeDocument21 pagesEnergies: An Optimum Enthalpy Approach For Melting and Solidification With Volume ChangeLuis Felipe Gutierrez MarcantoniNo ratings yet
- Thermo ChemistryDocument20 pagesThermo ChemistryTsabit AlbananiNo ratings yet
- Experiment 6 CalorimetryDocument4 pagesExperiment 6 CalorimetryJoshua EgallaNo ratings yet
- Calorimetry: 2.1 Measuring HeatDocument16 pagesCalorimetry: 2.1 Measuring HeatAnastasia Billin100% (1)
- Experiment 2. Vapor Pressure.Document4 pagesExperiment 2. Vapor Pressure.Esmeralda A OcampoNo ratings yet
- Example 3Document12 pagesExample 3girlhiNo ratings yet
- Physical Chemistry LabDocument11 pagesPhysical Chemistry LabBreyonnaMorganNo ratings yet
- Part 3Document25 pagesPart 3abdullahghaya124No ratings yet
- Exp06 WinDocument16 pagesExp06 Winmorumonday84No ratings yet
- Informe CarbajalDocument24 pagesInforme CarbajalshirleyNo ratings yet
- MT1 Saturday Review Session Solutions Phys7BDocument8 pagesMT1 Saturday Review Session Solutions Phys7BPatrickHindenburgNo ratings yet
- Silo - Tips 05 Enthalpy of Hydration of Sodium AcetateDocument7 pagesSilo - Tips 05 Enthalpy of Hydration of Sodium AcetateMarco MorelliNo ratings yet
- 0 00 4Document6 pages0 00 4Phong Sư LinhNo ratings yet
- Lecture 3 - Systems (SFEE) Specific Heats The Working FluidDocument38 pagesLecture 3 - Systems (SFEE) Specific Heats The Working FluidWillie MojataleNo ratings yet
- The ClausiusDocument12 pagesThe ClausiusjokishNo ratings yet
- Thermodynamics: Lecture 2 Review of Lecture 1Document10 pagesThermodynamics: Lecture 2 Review of Lecture 1Lexter Gomez GabicaNo ratings yet
- Non Ideal Gas TurbinesDocument4 pagesNon Ideal Gas TurbinesShaurya SachdevNo ratings yet
- E07 Hesslaw2016 PDFDocument8 pagesE07 Hesslaw2016 PDFKartikNo ratings yet
- Thermodynamics I: 1 Ideal Gases and Heat EnginesDocument18 pagesThermodynamics I: 1 Ideal Gases and Heat EnginesT.S GoriNo ratings yet
- 2012EJP ClausiusClapeyron CorrectedDocument11 pages2012EJP ClausiusClapeyron CorrectedderyhermawanNo ratings yet
- Thermodynamics I: 1 Ideal Gases and Heat EnginesDocument18 pagesThermodynamics I: 1 Ideal Gases and Heat Engineswixidi6042No ratings yet
- Combined Gas LawDocument17 pagesCombined Gas LawChesterJeorgeDizonNo ratings yet
- Pure SubstancesDocument36 pagesPure SubstancespavanraneNo ratings yet
- Lumped Capacitance ExperimentDocument10 pagesLumped Capacitance ExperimentDorian GreyNo ratings yet
- Correction TempDocument22 pagesCorrection TempFahmi BajryNo ratings yet
- Thermodynamics Progate: Rituparnsomvanshi May 2019Document29 pagesThermodynamics Progate: Rituparnsomvanshi May 2019Rituparn SinghNo ratings yet
- Lect 1Document14 pagesLect 1nagaraj108100% (1)
- Concentric Tube Heat ExchangersDocument8 pagesConcentric Tube Heat ExchangersHazryNo ratings yet
- Mid1 PhuongLT 20201690Document5 pagesMid1 PhuongLT 20201690dothuan20112001No ratings yet
- Clausius-Clapeyron Equation and Saturation Vapour Pressure: Simple Theory Reconciled With PracticeDocument11 pagesClausius-Clapeyron Equation and Saturation Vapour Pressure: Simple Theory Reconciled With Practicemutmainnah innaNo ratings yet
- Process Modelling, Simulation and Control For Chemical Engineering. Solved Problems. Chapter 5: Simulation Ex-AmplesDocument12 pagesProcess Modelling, Simulation and Control For Chemical Engineering. Solved Problems. Chapter 5: Simulation Ex-AmplesJohn100% (2)
- Determination of The Thermal PropertiesDocument10 pagesDetermination of The Thermal PropertiesPərviz HacızadəNo ratings yet
- Problem Set 10 Key - Physical Chemistry For Engineers. (Book Work)Document8 pagesProblem Set 10 Key - Physical Chemistry For Engineers. (Book Work)krymxenNo ratings yet
- Heat Transfer IIDocument13 pagesHeat Transfer IIpasha khanNo ratings yet
- Thermal Physics Lecture 22Document7 pagesThermal Physics Lecture 22OmegaUserNo ratings yet
- Chemistry Discipline Khulna UniversityDocument4 pagesChemistry Discipline Khulna Universityমেরাজ সিএস চৌধুরীNo ratings yet
- Entropy Changes & Processes Entropy at A Phase TransitionDocument4 pagesEntropy Changes & Processes Entropy at A Phase TransitionnabilanftNo ratings yet
- Heat of Fusion of WaterDocument6 pagesHeat of Fusion of WaterAishaNo ratings yet
- SCHX1014 - Chemical Engineering Thermodynamics - Unit 3Document17 pagesSCHX1014 - Chemical Engineering Thermodynamics - Unit 3Shanmuga PriyaNo ratings yet
- Thermodynamics ParadoxesDocument20 pagesThermodynamics Paradoxesmaria fernandaNo ratings yet
- CalorimetryDocument10 pagesCalorimetryDaizLee Ahmad0% (1)
- ΔHmix 2Document11 pagesΔHmix 2Nohan JoemonNo ratings yet
- A Modern Course in Statistical PhysicsFrom EverandA Modern Course in Statistical PhysicsRating: 3.5 out of 5 stars3.5/5 (2)
- Local Grid RefinementDocument10 pagesLocal Grid RefinementWallace StationNo ratings yet
- (DPP-4) - (JEE 2.0) - Basic Mathematics - Inverse Trigonometric Functions - 18th MayDocument47 pages(DPP-4) - (JEE 2.0) - Basic Mathematics - Inverse Trigonometric Functions - 18th MayAmit TimalsinaNo ratings yet
- Model Question Paper Subject Code: MC0068 Subject Name: Data Structure Using C Credits: 4 Marks: 140 Part A (One Mark Questions)Document13 pagesModel Question Paper Subject Code: MC0068 Subject Name: Data Structure Using C Credits: 4 Marks: 140 Part A (One Mark Questions)sailendra31No ratings yet
- CSO Gaddis Java Chapter03 6eDocument63 pagesCSO Gaddis Java Chapter03 6eMuhamad IhsanNo ratings yet
- Olymon2010 Mathematical Olympiads Correspondence ProgramDocument35 pagesOlymon2010 Mathematical Olympiads Correspondence ProgramOscar Reynaga AlarcónNo ratings yet
- Mathematics of BookmakingDocument10 pagesMathematics of BookmakingGiorgi BalkhamishviliNo ratings yet
- AI Project CycleDocument74 pagesAI Project Cycleashmita11No ratings yet
- 3rd Quarterly Exam in Business MathDocument1 page3rd Quarterly Exam in Business MathDaizy Alarcon AvilaNo ratings yet
- Notes From Anton 10 TH Ed CH 4Document5 pagesNotes From Anton 10 TH Ed CH 4Web MastersNo ratings yet
- Static Analysis of Slug FlowDocument8 pagesStatic Analysis of Slug FlowHmd MokhtariNo ratings yet
- Epa-1664 (Oil and Grease)Document28 pagesEpa-1664 (Oil and Grease)iqciro0% (1)
- Chapter 1Document87 pagesChapter 1nikhilNo ratings yet
- 30 Case Study Nakheel Tower The Vertical CityDocument8 pages30 Case Study Nakheel Tower The Vertical CityAR UmairNo ratings yet
- Sorting Algorithm in C-Bubble Sort and Selection SortDocument30 pagesSorting Algorithm in C-Bubble Sort and Selection SortShrishail KambleNo ratings yet
- Consumer Choice: X, Y X Y XDocument4 pagesConsumer Choice: X, Y X Y XBrianNo ratings yet
- Problem P4.17: The Trough of Concrete Weighs 800 Lb. (A) Draw A Free Body Diagram of TheDocument4 pagesProblem P4.17: The Trough of Concrete Weighs 800 Lb. (A) Draw A Free Body Diagram of TheKurtNo ratings yet
- Maritime Books by Indian Authors-Part I PDFDocument2 pagesMaritime Books by Indian Authors-Part I PDFTusshar Jaiswal100% (1)
- CH07 Characters, Strings and StringBuilderDocument43 pagesCH07 Characters, Strings and StringBuilderNhemi EstradaNo ratings yet
- Packed N Fluidized BedDocument6 pagesPacked N Fluidized BedhariprakavNo ratings yet
- Streamline SimulationDocument48 pagesStreamline SimulationAnjan KumarNo ratings yet
- ROBOTC Curriculum Outline PDFDocument73 pagesROBOTC Curriculum Outline PDFsalborunda100% (1)
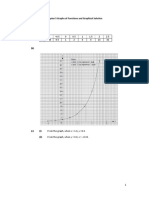
- Chapter 5 Graphs of Functions and Graphical Solution Exercise 5B 1. (A) 4 X - 1 - 0.5 0 0.5 1 1.5 2 2.5 y 0.25 0.5 1 2 4 8 16 32 (B)Document8 pagesChapter 5 Graphs of Functions and Graphical Solution Exercise 5B 1. (A) 4 X - 1 - 0.5 0 0.5 1 1.5 2 2.5 y 0.25 0.5 1 2 4 8 16 32 (B)Ashley HongNo ratings yet
- Ansi C119.4 (1998)Document23 pagesAnsi C119.4 (1998)juanita sanchez buitragoNo ratings yet
- Assignment - 2 - Electric Charges and Fields-Questions PDFDocument9 pagesAssignment - 2 - Electric Charges and Fields-Questions PDFvrajmenon6260No ratings yet
- PresentationDocument33 pagesPresentationYoftahiNo ratings yet
- 12 Unit - 2 PPT Oscillations and Waves 2008Document221 pages12 Unit - 2 PPT Oscillations and Waves 2008tsegab bekele0% (1)
- 03 TCAD Laboratory Overview of Synopsys Sentaurus TCAD GBB FinalAA13-14Document31 pages03 TCAD Laboratory Overview of Synopsys Sentaurus TCAD GBB FinalAA13-14vpsampathNo ratings yet
- Juan Ojeda and Nelson Hern AndezDocument6 pagesJuan Ojeda and Nelson Hern AndezNelson HernandezNo ratings yet