
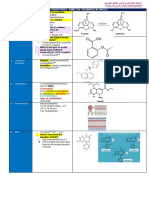
Unit 5 Industrial Pharmacy 2 7th Sem
Unit 5 Industrial Pharmacy 2 7th Sem
You might also like
- Lesson Plan ContentDocument6 pagesLesson Plan ContentSrijana Gurung100% (2)
- StructuresDocument7 pagesStructuresFarouk Ramadan100% (1)
- Regulatory Affairs PDFDocument27 pagesRegulatory Affairs PDFDipak BhingardeveNo ratings yet
- IVT Network - The FDA CGMP Inspection Is Coming - Make The Best of It - 2016-02-05Document18 pagesIVT Network - The FDA CGMP Inspection Is Coming - Make The Best of It - 2016-02-05marwaNo ratings yet
- Psychopharmacology LectureDocument29 pagesPsychopharmacology LectureTarek Qawasmeh100% (3)
- BP702T Ip VDocument29 pagesBP702T Ip VMilind JagdaleNo ratings yet
- Regulatory AspectsDocument42 pagesRegulatory Aspectspiyusharora1964No ratings yet
- Ipp 2 PPT .1Document24 pagesIpp 2 PPT .1mamta maliNo ratings yet
- Industrial Pharmacy II Unit 5Document29 pagesIndustrial Pharmacy II Unit 5Karan PathakNo ratings yet
- IP - Unit 5Document27 pagesIP - Unit 5Aditya PrajapatiNo ratings yet
- Cdsco: Presentation byDocument12 pagesCdsco: Presentation bymeenu sruthi priyaNo ratings yet
- Regulatory Affairs PDFDocument12 pagesRegulatory Affairs PDFdrgdsw50% (2)
- Juris NepalDocument8 pagesJuris NepalAmrit GaireNo ratings yet
- Resources For IND Applications: Back To TopDocument4 pagesResources For IND Applications: Back To Topkavya nainitaNo ratings yet
- What Is FDADocument10 pagesWhat Is FDAAchuthanand MukundanNo ratings yet
- Unit 2 Regulatory Authorities: StructureDocument18 pagesUnit 2 Regulatory Authorities: Structurevikas__ccNo ratings yet
- 05 Industrial Pharmacy II Unit VDocument15 pages05 Industrial Pharmacy II Unit VRahul VermaNo ratings yet
- Clinical Trials in IndiaDocument3 pagesClinical Trials in Indiawubbalubbadubdub200475No ratings yet
- Prelim PrpiDocument118 pagesPrelim PrpiShannen CostoNo ratings yet
- PRESENTATION On Drug Regulatory Authority On IndiaDocument23 pagesPRESENTATION On Drug Regulatory Authority On Indiaimrajansingh80No ratings yet
- Lecture # 8 Dr. Laiq (6.10.19) PDFDocument50 pagesLecture # 8 Dr. Laiq (6.10.19) PDFAbbas HassanNo ratings yet
- CRCP Lecture Reg Approvals Oct 2020Document65 pagesCRCP Lecture Reg Approvals Oct 2020EsEnGauharNo ratings yet
- The FDA Export Registration Requirements: 2. General Export Requirements Under Export Reform and Enhancement ActDocument4 pagesThe FDA Export Registration Requirements: 2. General Export Requirements Under Export Reform and Enhancement ActDeepika BairageeNo ratings yet
- 1 Indndaandasnda-210220130515Document21 pages1 Indndaandasnda-210220130515DeepaNo ratings yet
- Anupama Ra NotesDocument23 pagesAnupama Ra NotesShahzeel Iftikhar100% (1)
- 06 - Regulatory EnvironmentDocument6 pages06 - Regulatory EnvironmentLaura SaglietiNo ratings yet
- Chapter 2Document11 pagesChapter 2Surround TechsNo ratings yet
- Module 2 - Regulatory Approval Process - Lecture NotesDocument16 pagesModule 2 - Regulatory Approval Process - Lecture NotesyvssmanjunathNo ratings yet
- Lecture 1 - IntroductionDocument11 pagesLecture 1 - IntroductionyahyaNo ratings yet
- MASS Pharmacy Law 2014Document313 pagesMASS Pharmacy Law 20147bostondrNo ratings yet
- Drugs Approving AuthoritiesDocument38 pagesDrugs Approving AuthoritiesTariq HaqueNo ratings yet
- GUIDELINE FOR REGISTRATION OF DRUG-MEDICAL DEVICE AND MEDICAL DEVICE-DRUG COMBINATION PRODUCTS 4th Edition - 6th October 2021Document60 pagesGUIDELINE FOR REGISTRATION OF DRUG-MEDICAL DEVICE AND MEDICAL DEVICE-DRUG COMBINATION PRODUCTS 4th Edition - 6th October 2021KS WongNo ratings yet
- 3 Combined Unit I and Unit II Part 3 Administration of The Acts & RulesDocument30 pages3 Combined Unit I and Unit II Part 3 Administration of The Acts & RulesmavushieditzNo ratings yet
- Veterinary BiologicalDocument124 pagesVeterinary BiologicalsatishNo ratings yet
- Regulatory RequirementsDocument6 pagesRegulatory RequirementsAmeeroddin Mohammad100% (1)
- Regulatory Requirements For Manufacturing, Import and New Drug Approval in IndiaDocument14 pagesRegulatory Requirements For Manufacturing, Import and New Drug Approval in IndiaLenisha SequeiraNo ratings yet
- Introduction To Drug Regulatory SystemDocument40 pagesIntroduction To Drug Regulatory Systemrv nidinNo ratings yet
- Regulatory Requirements For Clinical TrialDocument8 pagesRegulatory Requirements For Clinical TrialGyana SahooNo ratings yet
- Regulatory Requirements For Drug Product ApprovalDocument1 pageRegulatory Requirements For Drug Product Approval51-Suryawanshi ShraddhaNo ratings yet
- Fda Human Drug Review and Approval Basics ModuleDocument8 pagesFda Human Drug Review and Approval Basics ModuleTawfeeq BA AbbadNo ratings yet
- Unit 5 Notes DRADocument14 pagesUnit 5 Notes DRASkb ArsalaanNo ratings yet
- Regulatory Issues in The Indian Pharmaceutical IndustryDocument20 pagesRegulatory Issues in The Indian Pharmaceutical IndustryrayyanNo ratings yet
- Regulatory Requirements For Product ApprovalDocument13 pagesRegulatory Requirements For Product Approval50KMKDIVYA RAJPALNo ratings yet
- USFDADocument69 pagesUSFDAtharunika deepaNo ratings yet
- DCGI Drive To Push Generics and PhaseDocument5 pagesDCGI Drive To Push Generics and PhaseRamchandra KenyNo ratings yet
- DrugscosmeticsactDocument70 pagesDrugscosmeticsacthemihemaNo ratings yet
- Guideline For Registration of Drug-Medical Device and Medical Device-Drug Combinations ProductsDocument33 pagesGuideline For Registration of Drug-Medical Device and Medical Device-Drug Combinations ProductsTZ LABNo ratings yet
- CAP-3-JULY-23--merged_1688399282Document40 pagesCAP-3-JULY-23--merged_1688399282iamashwinidubeyNo ratings yet
- GMP For Facility Design References April06Document17 pagesGMP For Facility Design References April06madhubiochemNo ratings yet
- Commentary On New Drugs and Cosmetics Bill 2022Document11 pagesCommentary On New Drugs and Cosmetics Bill 2022PracheekNo ratings yet
- GPA-Approach To ChinaDocument7 pagesGPA-Approach To ChinaGuille PBNo ratings yet
- Inhaler Registration - Drug-MD GuidelineDocument33 pagesInhaler Registration - Drug-MD Guidelinetan_hoe_1No ratings yet
- Frequently Asked Questions (Faqs) On New Drugs and Clinical TrialsDocument34 pagesFrequently Asked Questions (Faqs) On New Drugs and Clinical TrialsdnalokeshNo ratings yet
- UNIT-3 2. Role of RADocument5 pagesUNIT-3 2. Role of RADheeraj JaiswalNo ratings yet
- Introduction to Drug Regulatory AffairsDocument8 pagesIntroduction to Drug Regulatory AffairsShabnam YadavNo ratings yet
- Srilaksmi 2017 Regulatory Requirements For Registration of API in US and EU PDFDocument17 pagesSrilaksmi 2017 Regulatory Requirements For Registration of API in US and EU PDFhira darNo ratings yet
- DMF PDFDocument17 pagesDMF PDFAl RammohanNo ratings yet
- Assignment ON: Critical Analysis of New Guidelines For Clinical Research OrganizationsDocument10 pagesAssignment ON: Critical Analysis of New Guidelines For Clinical Research Organizationsdeepsonu15685No ratings yet
- Global Regulatory Agencies & FDA TrendsDocument29 pagesGlobal Regulatory Agencies & FDA TrendsUma MaheswariNo ratings yet
- CDSCO and State Licensing AuthorityDocument14 pagesCDSCO and State Licensing AuthorityPAREKH DARSHANNo ratings yet
- New SeminarDocument15 pagesNew Seminarolanrewaju moshoodNo ratings yet
- Overview of Drug Regulatory Affairs and Regulatory ProfessionDocument4 pagesOverview of Drug Regulatory Affairs and Regulatory ProfessionPriyank VariavaNo ratings yet
- The FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished PharmaceuticalsFrom EverandThe FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished PharmaceuticalsNo ratings yet
- Adobe Scan Apr 01, 2022Document7 pagesAdobe Scan Apr 01, 2022Durgha SureshNo ratings yet
- Adobe Scan 01 Oct 2020Document2 pagesAdobe Scan 01 Oct 2020Durgha SureshNo ratings yet
- Unit 2 Industrial Pharmacy 2 7th SemesterDocument29 pagesUnit 2 Industrial Pharmacy 2 7th SemesterDurgha SureshNo ratings yet
- Unit 3 Industrial Pharmacy 2 7th SemesterDocument27 pagesUnit 3 Industrial Pharmacy 2 7th SemesterDurgha SureshNo ratings yet
- VaginoplastyDocument16 pagesVaginoplastyAmanNo ratings yet
- List of Abbreviations Used in Medical PrescriptionsDocument7 pagesList of Abbreviations Used in Medical PrescriptionsZulaikah Nur IstiqomahNo ratings yet
- Elisabeth Johanne Rook: Curriculum VitaeDocument2 pagesElisabeth Johanne Rook: Curriculum VitaeAbdullah TheNo ratings yet
- Open Comparative Study of Efficacy and Safety of Ketoconazole Soap and Oral Ketoconazole in Tinea VersicolorDocument5 pagesOpen Comparative Study of Efficacy and Safety of Ketoconazole Soap and Oral Ketoconazole in Tinea VersicolorYohanes WidjajaNo ratings yet
- Facts About Abilify Maintena: (Aripiprazole) For Extended-Release Injectable Suspension in Patients With SchizophreniaDocument4 pagesFacts About Abilify Maintena: (Aripiprazole) For Extended-Release Injectable Suspension in Patients With SchizophreniarajeevsonaliNo ratings yet
- Pharmacology PrescriptionsDocument6 pagesPharmacology PrescriptionsJohn PayneNo ratings yet
- Drug Dosing ChartDocument16 pagesDrug Dosing ChartdnaaziiNo ratings yet
- HIV Infection Case StudiesDocument12 pagesHIV Infection Case StudiesAmnah HashmiNo ratings yet
- Augmentin: Brand Name: Generic Name: Drug ClassificationDocument2 pagesAugmentin: Brand Name: Generic Name: Drug ClassificationChristine Pialan SalimbagatNo ratings yet
- Listade PreciosDocument52 pagesListade PreciosEligio SalasNo ratings yet
- Analisis Waktu Tunggu Pelayanan Resep Di Puskesmas Pasir Panjang Kota Kupang Bulan April Tahun 2018Document8 pagesAnalisis Waktu Tunggu Pelayanan Resep Di Puskesmas Pasir Panjang Kota Kupang Bulan April Tahun 2018Ridhatul AzizahNo ratings yet
- Apollo PharmacyDocument17 pagesApollo PharmacyPrateek GargNo ratings yet
- Optimizing Real-World Benefit and Risk of New Psychedelic Medications - The Need For Innovative Postmarket SurveillanceDocument9 pagesOptimizing Real-World Benefit and Risk of New Psychedelic Medications - The Need For Innovative Postmarket Surveillance9newsNo ratings yet
- 68922Document40 pages68922arnidaNo ratings yet
- Medication Dialogue: Obs. in Loc de How Are You Feeling?", Putem Folosi How Do You Feel?", Fara Sa SchimbamDocument4 pagesMedication Dialogue: Obs. in Loc de How Are You Feeling?", Putem Folosi How Do You Feel?", Fara Sa SchimbamToma GeorgianaaNo ratings yet
- DRUG STUDY - AnticonvulsantsDocument1 pageDRUG STUDY - AnticonvulsantsZam PamateNo ratings yet
- Drug Study MetforminDocument5 pagesDrug Study MetforminSabita PaudelNo ratings yet
- PERTEMUAN 2. Kompartemen 1 TerbukaDocument50 pagesPERTEMUAN 2. Kompartemen 1 TerbukaKusuma WardaniNo ratings yet
- National Essential Drugs ListDocument50 pagesNational Essential Drugs ListportosinNo ratings yet
- Daftar BukuDocument30 pagesDaftar BukuRezeki AdnaveNo ratings yet
- HEM 53 Timetable 2012 Ver 3Document8 pagesHEM 53 Timetable 2012 Ver 3Chema SaizNo ratings yet
- Creative AdjustmentDocument56 pagesCreative AdjustmentNancy Azanasía Karantzia100% (2)
- SKM C454e15081914060Document1 pageSKM C454e15081914060Timmy NguyenNo ratings yet
- Biopharmaceutical Considerations in Drug Product Design and in Vitro Introduction (Biopharm)Document22 pagesBiopharmaceutical Considerations in Drug Product Design and in Vitro Introduction (Biopharm)vipinkv99No ratings yet
- Side Effects of Anti Psychotic MedicationsDocument11 pagesSide Effects of Anti Psychotic MedicationsluciapopNo ratings yet
- Daftar Obat High AlertDocument31 pagesDaftar Obat High Alertmuin ritongaNo ratings yet
- AntidepressantsDocument31 pagesAntidepressantsIzhaan AkmalNo ratings yet




























































































Download as pdf or txt
You might also like
- Lesson Plan ContentDocument6 pagesLesson Plan ContentSrijana Gurung100% (2)
- StructuresDocument7 pagesStructuresFarouk Ramadan100% (1)
- Regulatory Affairs PDFDocument27 pagesRegulatory Affairs PDFDipak BhingardeveNo ratings yet
- IVT Network - The FDA CGMP Inspection Is Coming - Make The Best of It - 2016-02-05Document18 pagesIVT Network - The FDA CGMP Inspection Is Coming - Make The Best of It - 2016-02-05marwaNo ratings yet
- Psychopharmacology LectureDocument29 pagesPsychopharmacology LectureTarek Qawasmeh100% (3)
- BP702T Ip VDocument29 pagesBP702T Ip VMilind JagdaleNo ratings yet
- Regulatory AspectsDocument42 pagesRegulatory Aspectspiyusharora1964No ratings yet
- Ipp 2 PPT .1Document24 pagesIpp 2 PPT .1mamta maliNo ratings yet
- Industrial Pharmacy II Unit 5Document29 pagesIndustrial Pharmacy II Unit 5Karan PathakNo ratings yet
- IP - Unit 5Document27 pagesIP - Unit 5Aditya PrajapatiNo ratings yet
- Cdsco: Presentation byDocument12 pagesCdsco: Presentation bymeenu sruthi priyaNo ratings yet
- Regulatory Affairs PDFDocument12 pagesRegulatory Affairs PDFdrgdsw50% (2)
- Juris NepalDocument8 pagesJuris NepalAmrit GaireNo ratings yet
- Resources For IND Applications: Back To TopDocument4 pagesResources For IND Applications: Back To Topkavya nainitaNo ratings yet
- What Is FDADocument10 pagesWhat Is FDAAchuthanand MukundanNo ratings yet
- Unit 2 Regulatory Authorities: StructureDocument18 pagesUnit 2 Regulatory Authorities: Structurevikas__ccNo ratings yet
- 05 Industrial Pharmacy II Unit VDocument15 pages05 Industrial Pharmacy II Unit VRahul VermaNo ratings yet
- Clinical Trials in IndiaDocument3 pagesClinical Trials in Indiawubbalubbadubdub200475No ratings yet
- Prelim PrpiDocument118 pagesPrelim PrpiShannen CostoNo ratings yet
- PRESENTATION On Drug Regulatory Authority On IndiaDocument23 pagesPRESENTATION On Drug Regulatory Authority On Indiaimrajansingh80No ratings yet
- Lecture # 8 Dr. Laiq (6.10.19) PDFDocument50 pagesLecture # 8 Dr. Laiq (6.10.19) PDFAbbas HassanNo ratings yet
- CRCP Lecture Reg Approvals Oct 2020Document65 pagesCRCP Lecture Reg Approvals Oct 2020EsEnGauharNo ratings yet
- The FDA Export Registration Requirements: 2. General Export Requirements Under Export Reform and Enhancement ActDocument4 pagesThe FDA Export Registration Requirements: 2. General Export Requirements Under Export Reform and Enhancement ActDeepika BairageeNo ratings yet
- 1 Indndaandasnda-210220130515Document21 pages1 Indndaandasnda-210220130515DeepaNo ratings yet
- Anupama Ra NotesDocument23 pagesAnupama Ra NotesShahzeel Iftikhar100% (1)
- 06 - Regulatory EnvironmentDocument6 pages06 - Regulatory EnvironmentLaura SaglietiNo ratings yet
- Chapter 2Document11 pagesChapter 2Surround TechsNo ratings yet
- Module 2 - Regulatory Approval Process - Lecture NotesDocument16 pagesModule 2 - Regulatory Approval Process - Lecture NotesyvssmanjunathNo ratings yet
- Lecture 1 - IntroductionDocument11 pagesLecture 1 - IntroductionyahyaNo ratings yet
- MASS Pharmacy Law 2014Document313 pagesMASS Pharmacy Law 20147bostondrNo ratings yet
- Drugs Approving AuthoritiesDocument38 pagesDrugs Approving AuthoritiesTariq HaqueNo ratings yet
- GUIDELINE FOR REGISTRATION OF DRUG-MEDICAL DEVICE AND MEDICAL DEVICE-DRUG COMBINATION PRODUCTS 4th Edition - 6th October 2021Document60 pagesGUIDELINE FOR REGISTRATION OF DRUG-MEDICAL DEVICE AND MEDICAL DEVICE-DRUG COMBINATION PRODUCTS 4th Edition - 6th October 2021KS WongNo ratings yet
- 3 Combined Unit I and Unit II Part 3 Administration of The Acts & RulesDocument30 pages3 Combined Unit I and Unit II Part 3 Administration of The Acts & RulesmavushieditzNo ratings yet
- Veterinary BiologicalDocument124 pagesVeterinary BiologicalsatishNo ratings yet
- Regulatory RequirementsDocument6 pagesRegulatory RequirementsAmeeroddin Mohammad100% (1)
- Regulatory Requirements For Manufacturing, Import and New Drug Approval in IndiaDocument14 pagesRegulatory Requirements For Manufacturing, Import and New Drug Approval in IndiaLenisha SequeiraNo ratings yet
- Introduction To Drug Regulatory SystemDocument40 pagesIntroduction To Drug Regulatory Systemrv nidinNo ratings yet
- Regulatory Requirements For Clinical TrialDocument8 pagesRegulatory Requirements For Clinical TrialGyana SahooNo ratings yet
- Regulatory Requirements For Drug Product ApprovalDocument1 pageRegulatory Requirements For Drug Product Approval51-Suryawanshi ShraddhaNo ratings yet
- Fda Human Drug Review and Approval Basics ModuleDocument8 pagesFda Human Drug Review and Approval Basics ModuleTawfeeq BA AbbadNo ratings yet
- Unit 5 Notes DRADocument14 pagesUnit 5 Notes DRASkb ArsalaanNo ratings yet
- Regulatory Issues in The Indian Pharmaceutical IndustryDocument20 pagesRegulatory Issues in The Indian Pharmaceutical IndustryrayyanNo ratings yet
- Regulatory Requirements For Product ApprovalDocument13 pagesRegulatory Requirements For Product Approval50KMKDIVYA RAJPALNo ratings yet
- USFDADocument69 pagesUSFDAtharunika deepaNo ratings yet
- DCGI Drive To Push Generics and PhaseDocument5 pagesDCGI Drive To Push Generics and PhaseRamchandra KenyNo ratings yet
- DrugscosmeticsactDocument70 pagesDrugscosmeticsacthemihemaNo ratings yet
- Guideline For Registration of Drug-Medical Device and Medical Device-Drug Combinations ProductsDocument33 pagesGuideline For Registration of Drug-Medical Device and Medical Device-Drug Combinations ProductsTZ LABNo ratings yet
- CAP-3-JULY-23--merged_1688399282Document40 pagesCAP-3-JULY-23--merged_1688399282iamashwinidubeyNo ratings yet
- GMP For Facility Design References April06Document17 pagesGMP For Facility Design References April06madhubiochemNo ratings yet
- Commentary On New Drugs and Cosmetics Bill 2022Document11 pagesCommentary On New Drugs and Cosmetics Bill 2022PracheekNo ratings yet
- GPA-Approach To ChinaDocument7 pagesGPA-Approach To ChinaGuille PBNo ratings yet
- Inhaler Registration - Drug-MD GuidelineDocument33 pagesInhaler Registration - Drug-MD Guidelinetan_hoe_1No ratings yet
- Frequently Asked Questions (Faqs) On New Drugs and Clinical TrialsDocument34 pagesFrequently Asked Questions (Faqs) On New Drugs and Clinical TrialsdnalokeshNo ratings yet
- UNIT-3 2. Role of RADocument5 pagesUNIT-3 2. Role of RADheeraj JaiswalNo ratings yet
- Introduction to Drug Regulatory AffairsDocument8 pagesIntroduction to Drug Regulatory AffairsShabnam YadavNo ratings yet
- Srilaksmi 2017 Regulatory Requirements For Registration of API in US and EU PDFDocument17 pagesSrilaksmi 2017 Regulatory Requirements For Registration of API in US and EU PDFhira darNo ratings yet
- DMF PDFDocument17 pagesDMF PDFAl RammohanNo ratings yet
- Assignment ON: Critical Analysis of New Guidelines For Clinical Research OrganizationsDocument10 pagesAssignment ON: Critical Analysis of New Guidelines For Clinical Research Organizationsdeepsonu15685No ratings yet
- Global Regulatory Agencies & FDA TrendsDocument29 pagesGlobal Regulatory Agencies & FDA TrendsUma MaheswariNo ratings yet
- CDSCO and State Licensing AuthorityDocument14 pagesCDSCO and State Licensing AuthorityPAREKH DARSHANNo ratings yet
- New SeminarDocument15 pagesNew Seminarolanrewaju moshoodNo ratings yet
- Overview of Drug Regulatory Affairs and Regulatory ProfessionDocument4 pagesOverview of Drug Regulatory Affairs and Regulatory ProfessionPriyank VariavaNo ratings yet
- The FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished PharmaceuticalsFrom EverandThe FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished PharmaceuticalsNo ratings yet
- Adobe Scan Apr 01, 2022Document7 pagesAdobe Scan Apr 01, 2022Durgha SureshNo ratings yet
- Adobe Scan 01 Oct 2020Document2 pagesAdobe Scan 01 Oct 2020Durgha SureshNo ratings yet
- Unit 2 Industrial Pharmacy 2 7th SemesterDocument29 pagesUnit 2 Industrial Pharmacy 2 7th SemesterDurgha SureshNo ratings yet
- Unit 3 Industrial Pharmacy 2 7th SemesterDocument27 pagesUnit 3 Industrial Pharmacy 2 7th SemesterDurgha SureshNo ratings yet
- VaginoplastyDocument16 pagesVaginoplastyAmanNo ratings yet
- List of Abbreviations Used in Medical PrescriptionsDocument7 pagesList of Abbreviations Used in Medical PrescriptionsZulaikah Nur IstiqomahNo ratings yet
- Elisabeth Johanne Rook: Curriculum VitaeDocument2 pagesElisabeth Johanne Rook: Curriculum VitaeAbdullah TheNo ratings yet
- Open Comparative Study of Efficacy and Safety of Ketoconazole Soap and Oral Ketoconazole in Tinea VersicolorDocument5 pagesOpen Comparative Study of Efficacy and Safety of Ketoconazole Soap and Oral Ketoconazole in Tinea VersicolorYohanes WidjajaNo ratings yet
- Facts About Abilify Maintena: (Aripiprazole) For Extended-Release Injectable Suspension in Patients With SchizophreniaDocument4 pagesFacts About Abilify Maintena: (Aripiprazole) For Extended-Release Injectable Suspension in Patients With SchizophreniarajeevsonaliNo ratings yet
- Pharmacology PrescriptionsDocument6 pagesPharmacology PrescriptionsJohn PayneNo ratings yet
- Drug Dosing ChartDocument16 pagesDrug Dosing ChartdnaaziiNo ratings yet
- HIV Infection Case StudiesDocument12 pagesHIV Infection Case StudiesAmnah HashmiNo ratings yet
- Augmentin: Brand Name: Generic Name: Drug ClassificationDocument2 pagesAugmentin: Brand Name: Generic Name: Drug ClassificationChristine Pialan SalimbagatNo ratings yet
- Listade PreciosDocument52 pagesListade PreciosEligio SalasNo ratings yet
- Analisis Waktu Tunggu Pelayanan Resep Di Puskesmas Pasir Panjang Kota Kupang Bulan April Tahun 2018Document8 pagesAnalisis Waktu Tunggu Pelayanan Resep Di Puskesmas Pasir Panjang Kota Kupang Bulan April Tahun 2018Ridhatul AzizahNo ratings yet
- Apollo PharmacyDocument17 pagesApollo PharmacyPrateek GargNo ratings yet
- Optimizing Real-World Benefit and Risk of New Psychedelic Medications - The Need For Innovative Postmarket SurveillanceDocument9 pagesOptimizing Real-World Benefit and Risk of New Psychedelic Medications - The Need For Innovative Postmarket Surveillance9newsNo ratings yet
- 68922Document40 pages68922arnidaNo ratings yet
- Medication Dialogue: Obs. in Loc de How Are You Feeling?", Putem Folosi How Do You Feel?", Fara Sa SchimbamDocument4 pagesMedication Dialogue: Obs. in Loc de How Are You Feeling?", Putem Folosi How Do You Feel?", Fara Sa SchimbamToma GeorgianaaNo ratings yet
- DRUG STUDY - AnticonvulsantsDocument1 pageDRUG STUDY - AnticonvulsantsZam PamateNo ratings yet
- Drug Study MetforminDocument5 pagesDrug Study MetforminSabita PaudelNo ratings yet
- PERTEMUAN 2. Kompartemen 1 TerbukaDocument50 pagesPERTEMUAN 2. Kompartemen 1 TerbukaKusuma WardaniNo ratings yet
- National Essential Drugs ListDocument50 pagesNational Essential Drugs ListportosinNo ratings yet
- Daftar BukuDocument30 pagesDaftar BukuRezeki AdnaveNo ratings yet
- HEM 53 Timetable 2012 Ver 3Document8 pagesHEM 53 Timetable 2012 Ver 3Chema SaizNo ratings yet
- Creative AdjustmentDocument56 pagesCreative AdjustmentNancy Azanasía Karantzia100% (2)
- SKM C454e15081914060Document1 pageSKM C454e15081914060Timmy NguyenNo ratings yet
- Biopharmaceutical Considerations in Drug Product Design and in Vitro Introduction (Biopharm)Document22 pagesBiopharmaceutical Considerations in Drug Product Design and in Vitro Introduction (Biopharm)vipinkv99No ratings yet
- Side Effects of Anti Psychotic MedicationsDocument11 pagesSide Effects of Anti Psychotic MedicationsluciapopNo ratings yet
- Daftar Obat High AlertDocument31 pagesDaftar Obat High Alertmuin ritongaNo ratings yet
- AntidepressantsDocument31 pagesAntidepressantsIzhaan AkmalNo ratings yet