Download as pdf or txt
You might also like
- (Main) Genetic Engineering Lab Report Part 2 - Group 2Document17 pages(Main) Genetic Engineering Lab Report Part 2 - Group 2catarina alexandriaNo ratings yet
- Dr. Qudsia YousafiDocument30 pagesDr. Qudsia YousafiMalaika RehmanNo ratings yet
- Protein3dstructureprediction 161109084221Document30 pagesProtein3dstructureprediction 161109084221Jhanvi SNo ratings yet
- Homology Modelling Notes PDFDocument30 pagesHomology Modelling Notes PDFkamleshNo ratings yet
- Comparative Protein Structure Modeling Using ModellerDocument47 pagesComparative Protein Structure Modeling Using ModellerAlexNo ratings yet
- Bioinformatics Notes - 17Bt54: Module - 4Document48 pagesBioinformatics Notes - 17Bt54: Module - 4Samuel PrinceNo ratings yet
- Pieper NucleicAcidsRes 2004Document6 pagesPieper NucleicAcidsRes 2004ctak2022No ratings yet
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- Homolgy ModelingDocument19 pagesHomolgy ModelingJainendra JainNo ratings yet
- Eswar MethodsMolBiol 2008Document25 pagesEswar MethodsMolBiol 2008huouinkyoumaNo ratings yet
- (Methods in Molecular Biology, 2231) Kazutaka Katoh - Multiple Sequence Alignment - Methods and Protocols-Humana (2020)Document322 pages(Methods in Molecular Biology, 2231) Kazutaka Katoh - Multiple Sequence Alignment - Methods and Protocols-Humana (2020)Diego AlmeidaNo ratings yet
- Homology Modeling: Ref: Structural Bioinformatics, P.E Bourne Molecular Modeling, FolkersDocument16 pagesHomology Modeling: Ref: Structural Bioinformatics, P.E Bourne Molecular Modeling, FolkersPallavi VermaNo ratings yet
- Protein Structure Prediction Using Homology ModelingDocument11 pagesProtein Structure Prediction Using Homology Modelingthelmamapangela64No ratings yet
- Multiple Sequence AlignmentDocument6 pagesMultiple Sequence AlignmentGeovani Gregorio Rincon NavasNo ratings yet
- HomolgyDocument21 pagesHomolgySatish SahuNo ratings yet
- Competitive Learning Neural NetworkDocument62 pagesCompetitive Learning Neural Networktoon townNo ratings yet
- A Study On Protein Sequence Clustering With OPTICSDocument6 pagesA Study On Protein Sequence Clustering With OPTICSJournal of ComputingNo ratings yet
- Re Comb 2003Document16 pagesRe Comb 2003mustafaNo ratings yet
- 04-Alinemiento Múltiple de SecuenciasDocument14 pages04-Alinemiento Múltiple de SecuenciasAlexander Ccahuana IgnacioNo ratings yet
- Homology Modeling, Also Known As Comparative Modeling ofDocument19 pagesHomology Modeling, Also Known As Comparative Modeling ofManoharNo ratings yet
- Bif501 AssDocument3 pagesBif501 Asssana 568aNo ratings yet
- Tertiary Structure Prediction Methods: Any Given Protein SequenceDocument29 pagesTertiary Structure Prediction Methods: Any Given Protein Sequencesurrender003No ratings yet
- Pre-Assessment QuestionsDocument18 pagesPre-Assessment Questionsk.sachinNo ratings yet
- Conclusions and Future WorkDocument12 pagesConclusions and Future WorkashishkkrNo ratings yet
- (Art) Meta-Cognitive Neural Network For Classification Problems in A Sequential Learning FrameworkDocument11 pages(Art) Meta-Cognitive Neural Network For Classification Problems in A Sequential Learning FrameworkPrissTinkNo ratings yet
- Proteins: Benchmarking Template Selection and Model Quality Assessment For High-Resolution Comparative ModelingDocument10 pagesProteins: Benchmarking Template Selection and Model Quality Assessment For High-Resolution Comparative ModelingLavanya Priya SathyanNo ratings yet
- Protein Modelling ToolDocument6 pagesProtein Modelling ToolShovana DeyNo ratings yet
- Comparison of FRF and Modal Methods For CombiningDocument20 pagesComparison of FRF and Modal Methods For CombiningCaio CesarNo ratings yet
- 10 21105 Joss 05057Document5 pages10 21105 Joss 05057Arpanet ArpanetNo ratings yet
- Praline 3Document18 pagesPraline 3CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Homologymodeling 150123025144 Conversion Gate01Document27 pagesHomologymodeling 150123025144 Conversion Gate01Jhanvi SNo ratings yet
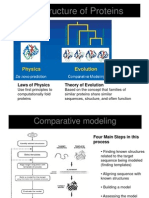
- 3-D Structure of Proteins: Laws of Physics Theory of EvolutionDocument9 pages3-D Structure of Proteins: Laws of Physics Theory of EvolutionladykikyoyunaNo ratings yet
- Metamotifs - A Generative Model For Building Families of Nucleotide Position Weight MatricesDocument16 pagesMetamotifs - A Generative Model For Building Families of Nucleotide Position Weight Matricesanon_174376942No ratings yet
- Protein Classification Using Hybrid Feature Selection TechniqueDocument9 pagesProtein Classification Using Hybrid Feature Selection TechniqueSudhakar TripathiNo ratings yet
- Supervised Machine Learning Techniques in Bioinformatics: Protein ClassificationDocument109 pagesSupervised Machine Learning Techniques in Bioinformatics: Protein ClassificationJoynul Abedin ZubairNo ratings yet
- Pi Is 0969212699801774Document14 pagesPi Is 0969212699801774Sahil8No ratings yet
- BIF101_II_Spring 2024 (1)Document8 pagesBIF101_II_Spring 2024 (1)My OpinionNo ratings yet
- tmp5573 TMPDocument11 pagestmp5573 TMPFrontiersNo ratings yet
- 1 s2.0 S136759312100051X MainDocument10 pages1 s2.0 S136759312100051X MainSumaiya Sadia BristyNo ratings yet
- Machine Learning For ChemistryDocument4 pagesMachine Learning For ChemistryLoga NathanNo ratings yet
- Unit 3Document9 pagesUnit 3document3580No ratings yet
- Vtu ML Lab ManualDocument47 pagesVtu ML Lab ManualNigar67% (3)
- ML Lab ManualDocument47 pagesML Lab Manualharshit gargNo ratings yet
- Homology ModellingDocument21 pagesHomology ModellingJerome FrancisNo ratings yet
- Protein ModellingDocument53 pagesProtein ModellingiluvgermieNo ratings yet
- Empirical Analysis of Performance Parameters For Consistency in Distributed DatabaseDocument29 pagesEmpirical Analysis of Performance Parameters For Consistency in Distributed DatabaseMona AbdelaleemNo ratings yet
- Unit-5 BioinformaticsDocument13 pagesUnit-5 Bioinformaticsp vmuraliNo ratings yet
- MsaDocument28 pagesMsaMausam KumravatNo ratings yet
- Blast 2 Sequences, A New Tool For Comparing Protein and Nucleotide SequencesDocument17 pagesBlast 2 Sequences, A New Tool For Comparing Protein and Nucleotide SequencesXâlmañ KháñNo ratings yet
- Comparison and Evaluation of Multiple Sequence Alignment Tools in BininformaticsDocument6 pagesComparison and Evaluation of Multiple Sequence Alignment Tools in Bininformaticsprashant_kaushal_2No ratings yet
- Sequence AnalysisDocument6 pagesSequence Analysisharrison9No ratings yet
- Effective Navigation of Query Results Based On Concept HierarchiesDocument17 pagesEffective Navigation of Query Results Based On Concept Hierarchiesakuthota6029No ratings yet
- 2013 J Theor Biol 328 77-88Document12 pages2013 J Theor Biol 328 77-88mbrylinskiNo ratings yet
- Theory: Sequence Alignment Is A Process of Aligning Two Sequences To Achieve Maximum Levels ofDocument5 pagesTheory: Sequence Alignment Is A Process of Aligning Two Sequences To Achieve Maximum Levels ofGopi ShankarNo ratings yet
- Lecture 101Document43 pagesLecture 101Jean ReneNo ratings yet
- Chapter 4 After ModfiyDocument4 pagesChapter 4 After Modfiyfatmahelawden000No ratings yet
- Machine Learning Laboratory SylbDocument1 pageMachine Learning Laboratory SylbVijay MahalingamNo ratings yet
- Combining Machine Learning and Agent-Based Modeling To Study Biomedical SystemsDocument33 pagesCombining Machine Learning and Agent-Based Modeling To Study Biomedical SystemsMarcelo Marcy Majstruk CimilloNo ratings yet
- DATA MINING AND MACHINE LEARNING. PREDICTIVE TECHNIQUES: REGRESSION, GENERALIZED LINEAR MODELS, SUPPORT VECTOR MACHINE AND NEURAL NETWORKSFrom EverandDATA MINING AND MACHINE LEARNING. PREDICTIVE TECHNIQUES: REGRESSION, GENERALIZED LINEAR MODELS, SUPPORT VECTOR MACHINE AND NEURAL NETWORKSNo ratings yet
- Class AssignmentDocument8 pagesClass AssignmentDishant kumar yadav mhakhariyaNo ratings yet
- Decision TreeDocument4 pagesDecision TreeDishant kumar yadav mhakhariyaNo ratings yet
- Lecture 9Document19 pagesLecture 9Dishant kumar yadav mhakhariyaNo ratings yet
- Lecture 12Document27 pagesLecture 12Dishant kumar yadav mhakhariyaNo ratings yet
- Lecture 13Document17 pagesLecture 13Dishant kumar yadav mhakhariyaNo ratings yet
- Lecture 15Document28 pagesLecture 15Dishant kumar yadav mhakhariyaNo ratings yet
- Lecture 2 - Barriers To CommunicationDocument11 pagesLecture 2 - Barriers To CommunicationDishant kumar yadav mhakhariyaNo ratings yet
- Kalasalingam Academy of Research and Education (Deemed To Be University) Anand Nagar, Krishnankoil - 626126Document60 pagesKalasalingam Academy of Research and Education (Deemed To Be University) Anand Nagar, Krishnankoil - 626126Dishant kumar yadav mhakhariyaNo ratings yet
- Lecture 3 - Non - Verbal CommunicationDocument26 pagesLecture 3 - Non - Verbal CommunicationDishant kumar yadav mhakhariyaNo ratings yet
- Certificate For COVID-19 Vaccination: Beneficiary DetailsDocument1 pageCertificate For COVID-19 Vaccination: Beneficiary DetailsNiranjan WarakeNo ratings yet
- Bacterialand Archael InclusionsDocument15 pagesBacterialand Archael InclusionsXimena AlcántaraNo ratings yet
- Laboratory Techniques in Rabies: Fourth EditionDocument494 pagesLaboratory Techniques in Rabies: Fourth EditionKimjhee Yang WongNo ratings yet
- Molecules of Life - TransesDocument3 pagesMolecules of Life - TransesJenny Ruth TubanNo ratings yet
- Enhanced Foreign Protein Accumulation in Nicotiana Benthamiana Leaves Co-Infiltrated With A TMV Vector and Plant Cell Cycle Regulator GenesDocument7 pagesEnhanced Foreign Protein Accumulation in Nicotiana Benthamiana Leaves Co-Infiltrated With A TMV Vector and Plant Cell Cycle Regulator Geneszhaoyue12112001No ratings yet
- Thesis For Research Paper On Genetic EngineeringDocument8 pagesThesis For Research Paper On Genetic Engineeringcnowkfhkf100% (1)
- Gmo Research Paper OutlineDocument7 pagesGmo Research Paper Outlinewtdcxtbnd100% (1)
- Li Et Al. 2022 - Untangling The Web of Intratumour HeterogeneityDocument10 pagesLi Et Al. 2022 - Untangling The Web of Intratumour HeterogeneitysodiogoesNo ratings yet
- Rivera-Laboratory Exercise No. 2-AnaphyDocument3 pagesRivera-Laboratory Exercise No. 2-AnaphyRivera, Trishia PilonNo ratings yet
- Nucleic Acid - An Overview - ScienceDirect TopicsDocument10 pagesNucleic Acid - An Overview - ScienceDirect TopicsBauza, Stephanie Dawn T.No ratings yet
- Flow Cytometry: Meroj A. JasemDocument58 pagesFlow Cytometry: Meroj A. Jasemahmad100% (1)
- Bms 07Document110 pagesBms 07Sabyasachi DasNo ratings yet
- Pseudo-Serendipity by Way of Random Variation: DNADocument7 pagesPseudo-Serendipity by Way of Random Variation: DNALeon LaupNo ratings yet
- Bio NotesDocument2 pagesBio NotesBobNo ratings yet
- The Discovery of CRISPR in Archaea and BacteriaDocument17 pagesThe Discovery of CRISPR in Archaea and BacteriaLightspeedNo ratings yet
- Cell Biology & BiochemistryDocument320 pagesCell Biology & BiochemistryVai SanNo ratings yet
- Red Blood Cell Membrane-Coated Functionalized Au Nanocage AsDocument12 pagesRed Blood Cell Membrane-Coated Functionalized Au Nanocage AsMuhammad Shehr YarNo ratings yet
- HIS-Select Technology GuideDocument6 pagesHIS-Select Technology GuideSigma-AldrichNo ratings yet
- Lab 8 - Transcription-Translation-ONLINE VERSION - 2021Document11 pagesLab 8 - Transcription-Translation-ONLINE VERSION - 2021thesoccerprince.10No ratings yet
- Alexander Et Al-2018-British Journal of Pharmacology PDFDocument113 pagesAlexander Et Al-2018-British Journal of Pharmacology PDFEduard José Martinez CamacaroNo ratings yet
- Purification of EnzymeDocument29 pagesPurification of Enzymeplastioid4079No ratings yet
- MIMM 211 MidtermDocument8 pagesMIMM 211 MidtermIpshitanandi100% (1)
- Mendel S Genes Molecular Characterization - 2011Document8 pagesMendel S Genes Molecular Characterization - 2011MARIA DE LOS ANGELES ORTIZ CRUZNo ratings yet
- G12 DR BiologyDocument196 pagesG12 DR BiologyAli RahimiNo ratings yet
- Post Graduate Degree Standard Paper - I Code:017Document2 pagesPost Graduate Degree Standard Paper - I Code:017Mk.jeyaNo ratings yet
- Neet Code C Question Paper PDFDocument41 pagesNeet Code C Question Paper PDFMagnifestoNo ratings yet
- Cell Resp and PhotosynDocument12 pagesCell Resp and Photosynhtb495No ratings yet
- Summer Training BrochureDocument1 pageSummer Training BrochureAbhilashRayaguruNo ratings yet
- Cell StructuresDocument54 pagesCell StructuresWilliam DC RiveraNo ratings yet