Download as pdf or txt
You might also like
- Performance Qualification Protocol and Report For AutoclaveDocument19 pagesPerformance Qualification Protocol and Report For AutoclaveMubarak PatelNo ratings yet
- Shadle PresentationDocument23 pagesShadle Presentationsherri_dobroskay4430100% (1)
- American National Standard For Filters Used inDocument14 pagesAmerican National Standard For Filters Used inBaskara PrabawaNo ratings yet
- Chromatography ColumnDocument8 pagesChromatography Columnyasa karyadaNo ratings yet
- cGMP's For Pharmaceutical Manufacturing: Priscilla AgiroDocument44 pagescGMP's For Pharmaceutical Manufacturing: Priscilla AgironuwaNo ratings yet
- 2013 Pharmaceutical Cleaning and Validation Alconox PresentationDocument74 pages2013 Pharmaceutical Cleaning and Validation Alconox PresentationMuhammad Tahir IqbalNo ratings yet
- Search Iso 8573Document2 pagesSearch Iso 8573Anonymous FZs3yBHh70% (1)
- CEN LSC AMER 5 Rules of Sensor Placement B211369EN ADocument11 pagesCEN LSC AMER 5 Rules of Sensor Placement B211369EN AmajaNo ratings yet
- Application Note: Successful Wetting For Filter Integrity Testing in Volume-Restricted SystemsDocument13 pagesApplication Note: Successful Wetting For Filter Integrity Testing in Volume-Restricted SystemsSlavaNo ratings yet
- FDA Warning Letter For Inadequate Batch Record ReviewDocument1 pageFDA Warning Letter For Inadequate Batch Record ReviewMina Maher MikhailNo ratings yet
- 04JA BlackburnDocument7 pages04JA BlackburnFederico BrigatoNo ratings yet
- 05JA ChvaicerDocument11 pages05JA ChvaiceramgranadosvNo ratings yet
- GUID - 6 en-USDocument37 pagesGUID - 6 en-USSantiago Cristancho100% (1)
- Ensuring The Air Suplly Rate To A Cleanroom Complies With The Eu GGMP and Iso 14644-3 Recovery Rate RequirementsDocument3 pagesEnsuring The Air Suplly Rate To A Cleanroom Complies With The Eu GGMP and Iso 14644-3 Recovery Rate RequirementsluisNo ratings yet
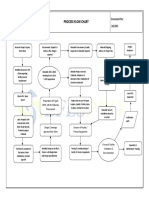
- Process Flow Chart NewDocument1 pageProcess Flow Chart NewSandhyaRamakrishnaNo ratings yet
- Filterintegritytesting-Sartorious Application NoteDocument8 pagesFilterintegritytesting-Sartorious Application NoteVijay Kumar NandagiriNo ratings yet
- Global Perspectives in Cleaning ValidationDocument17 pagesGlobal Perspectives in Cleaning ValidationYusuf SalahamanaNo ratings yet
- Fundamentals of Cleaning and Disinfection Programs For Aseptic Manufacturing FacilitiesDocument52 pagesFundamentals of Cleaning and Disinfection Programs For Aseptic Manufacturing FacilitiesKraken UrNo ratings yet
- Using A PQRI Approach in Process ValidationDocument18 pagesUsing A PQRI Approach in Process ValidationmthilekkumarNo ratings yet
- Cleanroom DesignDocument10 pagesCleanroom DesignshwampaNo ratings yet
- Methods For Testing Cleanroom GarmentsDocument7 pagesMethods For Testing Cleanroom GarmentsMINGZILANo ratings yet
- PVDocument52 pagesPVanjana sinhaNo ratings yet
- WP Gmp-En AnshDocument12 pagesWP Gmp-En AnshFelix ShihNo ratings yet
- Guidelines For Routine Environmental Cleaning of The Operating RoomDocument13 pagesGuidelines For Routine Environmental Cleaning of The Operating RoomYnaffit Alteza UntalNo ratings yet
- Smoke STUDIES ARTICLE 2 25 15 PDFDocument7 pagesSmoke STUDIES ARTICLE 2 25 15 PDFschumonNo ratings yet
- Air Handling UnitDocument3 pagesAir Handling UnitPrince Moni100% (2)
- Ausdiagnostics High-Plex Analyser Maintenance Procedure: 1. Standard Dtprime Maintenance GuideDocument12 pagesAusdiagnostics High-Plex Analyser Maintenance Procedure: 1. Standard Dtprime Maintenance GuideJimNo ratings yet
- GU - Routine Testing of Lab Balances - ENDocument8 pagesGU - Routine Testing of Lab Balances - ENsyifaurrohmah100% (1)
- IVT Network - 4 Indispensable Pre-Inspection Actions - 2014-02-27Document2 pagesIVT Network - 4 Indispensable Pre-Inspection Actions - 2014-02-27Mohammed YousffiNo ratings yet
- PHSS Annual Members Conference 2014: Book Online Phss - Co.uk/eventsDocument3 pagesPHSS Annual Members Conference 2014: Book Online Phss - Co.uk/eventsTim SandleNo ratings yet
- Who Inspection Hormone Product Manufacturing Facilities 2008 PDFDocument15 pagesWho Inspection Hormone Product Manufacturing Facilities 2008 PDFphamuyenthuNo ratings yet
- Three Tiers of Medical Device Process Validation Plans: Yeong-Lin ChenDocument16 pagesThree Tiers of Medical Device Process Validation Plans: Yeong-Lin ChengeorgesharmokhNo ratings yet
- Phuong - DryheatDocument5 pagesPhuong - DryheatqhpuongNo ratings yet
- Pe 009 17 GMP Guide XannexesDocument283 pagesPe 009 17 GMP Guide XannexesZeratul Chao0% (1)
- Who Inspection Hormone Product Manufacturing Facilities 2008Document15 pagesWho Inspection Hormone Product Manufacturing Facilities 2008Mostafa AfifyNo ratings yet
- FDA Inspections PDFDocument52 pagesFDA Inspections PDFMajdi Hasan Ayoub100% (1)
- Annex 1 Comments by Nissan CohenDocument15 pagesAnnex 1 Comments by Nissan CohenNarasimharao100% (1)
- A Practical Approach To Steam Autoclave Cycle Development: Byvictor Tsui, P.E. and Ward Wiederhold, B.S.M.EDocument10 pagesA Practical Approach To Steam Autoclave Cycle Development: Byvictor Tsui, P.E. and Ward Wiederhold, B.S.M.EJawad HussainNo ratings yet
- IspeDocument15 pagesIspeaneesh100No ratings yet
- Whythe10 ppmCriterionShouldBeAbandonedDocument5 pagesWhythe10 ppmCriterionShouldBeAbandonedMuhammad AsifNo ratings yet
- Preface Prologue Validation Master Plan Approval Page 1: 1.1 Project DescriptionDocument5 pagesPreface Prologue Validation Master Plan Approval Page 1: 1.1 Project DescriptionKyungSooLee100% (1)
- Bioproc PDFDocument6 pagesBioproc PDFSrijit KhanNo ratings yet
- 1 Water (Sumana)Document282 pages1 Water (Sumana)siruslara6491No ratings yet
- Usp 797GCDocument61 pagesUsp 797GCAwni1989No ratings yet
- Introduction To Science and Risk Based Cleaning Validation Using ASTM E3106 E3219Document9 pagesIntroduction To Science and Risk Based Cleaning Validation Using ASTM E3106 E3219nsk79inNo ratings yet
- Article2-Clean Operation Manufacturing294002107513883542Document4 pagesArticle2-Clean Operation Manufacturing294002107513883542авдей александрNo ratings yet
- Aseptic Processing White PaperDocument3 pagesAseptic Processing White PapermishannakNo ratings yet
- HEPA and ULPA Filters: The Aseptic CoreDocument7 pagesHEPA and ULPA Filters: The Aseptic CoreVaidhyanadhan DeepakNo ratings yet
- ISPE Print PageDocument1 pageISPE Print Pagesmallik3No ratings yet
- Process Validation of Polyherbal Cough Syrup FormulationDocument7 pagesProcess Validation of Polyherbal Cough Syrup FormulationBhavesh NayakNo ratings yet
- The Future of Systems Validation ACF1D6Document3 pagesThe Future of Systems Validation ACF1D6Mitchel JammalNo ratings yet
- ISO 21501-4 Perspectiva MetrológicaDocument8 pagesISO 21501-4 Perspectiva MetrológicaNicolas VargasNo ratings yet
- Isolator ISPE Study 2008Document51 pagesIsolator ISPE Study 2008edsonleviNo ratings yet
- Giz2012 en Comparison of Eu GMP Guidelines With Who Guidelines PDFDocument70 pagesGiz2012 en Comparison of Eu GMP Guidelines With Who Guidelines PDFMr ThanhNo ratings yet
- One 2 One Cross Contamination FinalDocument9 pagesOne 2 One Cross Contamination FinalBlank BacktobasicNo ratings yet
- Presentation - NEBBDocument7 pagesPresentation - NEBBBhavik Thakar100% (1)
- Data Integrity AwarenessDocument12 pagesData Integrity AwarenessPharmaceutical Guide100% (1)
- BOSCH Bio DecontaminationH2O2Document25 pagesBOSCH Bio DecontaminationH2O2Davide GrioniNo ratings yet
- Checklist - ISO 14001 - 2015Document9 pagesChecklist - ISO 14001 - 2015Rubini Devi SelvarajooNo ratings yet
- A. Read. Fill in The Blanks. (6 Marks) The Hill Some Buns A Bag Much FunDocument7 pagesA. Read. Fill in The Blanks. (6 Marks) The Hill Some Buns A Bag Much FunRubini Devi SelvarajooNo ratings yet
- Updated Tut Ch9B-2 T1-0 (Answer For Student)Document7 pagesUpdated Tut Ch9B-2 T1-0 (Answer For Student)Rubini Devi SelvarajooNo ratings yet
- Conversion FactorsDocument1 pageConversion FactorsRubini Devi SelvarajooNo ratings yet
- Sample For Laboratory Testing by Ls Pro Medan For Indonesia Sni ApplicationDocument2 pagesSample For Laboratory Testing by Ls Pro Medan For Indonesia Sni ApplicationRubini Devi SelvarajooNo ratings yet
- Ballast Water Treatment SystemDocument3 pagesBallast Water Treatment SystemNicole Audrey BusaNo ratings yet
- Bioethics and Biosafety in BiotechnologyDocument148 pagesBioethics and Biosafety in BiotechnologyPratibha Batra75% (4)
- Conclusion: (Citation Emb75 /L 1033)Document3 pagesConclusion: (Citation Emb75 /L 1033)Liyana HalimNo ratings yet
- Compact Air Drive Ii: User'S ManualDocument20 pagesCompact Air Drive Ii: User'S ManualrossiNo ratings yet
- Rapid Sterility Testing Using PallchekDocument29 pagesRapid Sterility Testing Using Pallchekvkumar6883No ratings yet
- Validation & Qualification of Medical DeviceDocument38 pagesValidation & Qualification of Medical DeviceMohammd Khush NoorNo ratings yet
- EMA 药品、活性物质、辅料和内包材灭菌指南-2019 (GMP办公室翻译组)Document27 pagesEMA 药品、活性物质、辅料和内包材灭菌指南-2019 (GMP办公室翻译组)于天一No ratings yet
- Sterisheet Focus N 1 PDF - 1372343937 62Document2 pagesSterisheet Focus N 1 PDF - 1372343937 62instrumed_globalNo ratings yet
- Glidescope Titanium Operations and Maintenance ManualDocument76 pagesGlidescope Titanium Operations and Maintenance ManualgrandmacaesarNo ratings yet
- Final HICC Manual AIIMSDocument35 pagesFinal HICC Manual AIIMSNaMakNo ratings yet
- Usp 2023 Microbiological Attributes of Nonsterile NutritionalDocument4 pagesUsp 2023 Microbiological Attributes of Nonsterile NutritionalChetalee NaikNo ratings yet
- Hairdressing SecondDocument46 pagesHairdressing SecondGracelyn GadorNo ratings yet
- Chemistry Processing Technology and Bio Energy PDFDocument334 pagesChemistry Processing Technology and Bio Energy PDFArvin SlayerNo ratings yet
- BKM-Z18-23B User Manual 2022.7.07-DesbloqueadoDocument42 pagesBKM-Z18-23B User Manual 2022.7.07-DesbloqueadoLuis GutierrezNo ratings yet
- Manual Rev-7 (05-2014)Document144 pagesManual Rev-7 (05-2014)Mohamed Choukri AzzoulaNo ratings yet
- Katalog Cartridge FilterDocument5 pagesKatalog Cartridge FilterJack AhmadNo ratings yet
- Compact Dry SL (For Salmonella) : 40/240/920 Plates Id No. 1002973/1002938/1002940Document1 pageCompact Dry SL (For Salmonella) : 40/240/920 Plates Id No. 1002973/1002938/1002940Wendy Núñez BedollaNo ratings yet
- RAPPLER - Ferrer Ferrer and Emmanuel - Incineration of Coronavirus Wastes Will Worsen The SituationDocument5 pagesRAPPLER - Ferrer Ferrer and Emmanuel - Incineration of Coronavirus Wastes Will Worsen The SituationRolando E. CaserNo ratings yet
- 12 Steps of Aseptic TechniqueDocument7 pages12 Steps of Aseptic TechniqueRobbie MejiaNo ratings yet
- Sterilisation of Water by Titration.Document8 pagesSterilisation of Water by Titration.Shivank SharmaNo ratings yet
- Kavya Nainita 19L81S0406 Ffs Technology Assignment PMTDocument8 pagesKavya Nainita 19L81S0406 Ffs Technology Assignment PMTkavya nainitaNo ratings yet
- (Psilocybin) Magic Mushrooms-A New Indoor Growing Technique (WWW - Erowid.org)Document29 pages(Psilocybin) Magic Mushrooms-A New Indoor Growing Technique (WWW - Erowid.org)Craig Ohlsen100% (7)
- USP 1231 - Water For Pharmaceutical PurposesDocument66 pagesUSP 1231 - Water For Pharmaceutical PurposesGiselle Clarisse D. CelizNo ratings yet
- SteriVac 8XL-Operator ManualDocument34 pagesSteriVac 8XL-Operator ManualHoang Nghiem Nghi100% (1)
- Whitepaperannex 1 TimsandleDocument45 pagesWhitepaperannex 1 TimsandleCalVal2006No ratings yet
- Activity 3 Aseptic Technique: Name: Cano, Claribelle V. Date: Laboratory Schedule: ScoreDocument3 pagesActivity 3 Aseptic Technique: Name: Cano, Claribelle V. Date: Laboratory Schedule: ScoreClaribelle CanoNo ratings yet
- Plasmatreat ImagebrochureDocument28 pagesPlasmatreat ImagebrochurechibigarNo ratings yet
- Laboratory Manual in General Microbiology For Undergraduate Students., ShortDocument49 pagesLaboratory Manual in General Microbiology For Undergraduate Students., ShortMaria AspriNo ratings yet
- Dry Fog BrochureDocument6 pagesDry Fog Brochuredani dumalangNo ratings yet