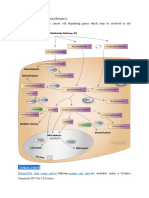
Download as pdf or txt
You might also like
- Uterine ProlapseDocument21 pagesUterine ProlapseFau Fau DheoboNo ratings yet
- Protein-Misfolding Diseases and Chaperone-Based Therapeutic ApproachesDocument19 pagesProtein-Misfolding Diseases and Chaperone-Based Therapeutic ApproachesTherese PagayNo ratings yet
- Glycolysis & Cancer Write UpDocument9 pagesGlycolysis & Cancer Write UpMarvin JeaNo ratings yet
- Protein Mutations in Disease: Lecture 11, Medical BiochemistryDocument27 pagesProtein Mutations in Disease: Lecture 11, Medical BiochemistrycafemedNo ratings yet
- The Role of p53 in Pancreatic Beta Cell ApoptosisDocument6 pagesThe Role of p53 in Pancreatic Beta Cell ApoptosisLuis Antonio FloresNo ratings yet
- Bergen Dahl 2019Document12 pagesBergen Dahl 2019Noor ShahizanNo ratings yet
- T10. Signaling in Control of Cell Growth and MetabolismDocument16 pagesT10. Signaling in Control of Cell Growth and MetabolismJavierNo ratings yet
- Fiskes&biokim Amenida Halawa-6120055Document15 pagesFiskes&biokim Amenida Halawa-6120055Festinidar GuloNo ratings yet
- Human Heredity Principles and Issues 10Th Edition Michael Cummings Solutions Manual Full Chapter PDFDocument32 pagesHuman Heredity Principles and Issues 10Th Edition Michael Cummings Solutions Manual Full Chapter PDFAshleyMaciasowbj100% (13)
- Human Heredity Principles and Issues 10th Edition Michael Cummings Solutions ManualDocument38 pagesHuman Heredity Principles and Issues 10th Edition Michael Cummings Solutions Manualbufochauswu2w100% (17)
- 1 s2.0 S1096719208000851 MainDocument4 pages1 s2.0 S1096719208000851 MainNermeen Mostafa ElhawaryNo ratings yet
- Chapter Protein Folding and MisfoldingDocument17 pagesChapter Protein Folding and MisfoldingMazhar FarNo ratings yet
- Bioinformatics Assignment: Medha Banerjee 1660091 IMTH-7 (A)Document11 pagesBioinformatics Assignment: Medha Banerjee 1660091 IMTH-7 (A)Medha BanerjeeNo ratings yet
- 483 Final HomeworkDocument6 pages483 Final HomeworkzeynoleeeNo ratings yet
- Ferroptosis: An Iron Dependent Cell Death Form Linking Metabolism, Diseases, Immune Cell and Targeted TherapyDocument12 pagesFerroptosis: An Iron Dependent Cell Death Form Linking Metabolism, Diseases, Immune Cell and Targeted Therapy14-120 RahmaNo ratings yet
- Full Human Heredity Principles and Issues 10Th Edition Michael Cummings Solutions Manual Online PDF All ChapterDocument34 pagesFull Human Heredity Principles and Issues 10Th Edition Michael Cummings Solutions Manual Online PDF All Chaptercflypsochewbfccf363100% (5)
- HypoxiaDocument8 pagesHypoxiabetelhem demekeNo ratings yet
- Ye 2019 Lipotoxicity and Beta Cell Maintenance in Obesity and Type 2 DiabetesDocument15 pagesYe 2019 Lipotoxicity and Beta Cell Maintenance in Obesity and Type 2 DiabetesPaul SimononNo ratings yet
- Nutrients 11 02923 v2Document18 pagesNutrients 11 02923 v2Manaloe90No ratings yet
- Apoptosis and CancerDocument4 pagesApoptosis and CancerhibaNo ratings yet
- AngelaDocument27 pagesAngelaMehedi HossainNo ratings yet
- Branched-Chain Amino Acid Supplementation: Impact On Signaling and Relevance To Critical IllnessDocument13 pagesBranched-Chain Amino Acid Supplementation: Impact On Signaling and Relevance To Critical IllnesssarahfajriaNo ratings yet
- Cancer Cell Metabolism - Cairns HarrisDocument14 pagesCancer Cell Metabolism - Cairns HarrisGUSTAVO BELLONo ratings yet
- Gamerdinger-2011-Emerging Roles of MoDocument8 pagesGamerdinger-2011-Emerging Roles of Motobias.hirnetNo ratings yet
- Methods in Molecular Biology, Vol.003 - New Protein TechniquesDocument524 pagesMethods in Molecular Biology, Vol.003 - New Protein TechniquesPablo HenrriquezNo ratings yet
- Cell Cycle and Cancer - Unit I IV/IDocument6 pagesCell Cycle and Cancer - Unit I IV/IManoj GourojuNo ratings yet
- Seminar On Protein and Peptides Drug DeliveryDocument37 pagesSeminar On Protein and Peptides Drug DeliverykeyurNo ratings yet
- Ifosfamide PathwayDocument6 pagesIfosfamide PathwayDiana AhmadNo ratings yet
- Food & Genes: Term Paper By: Phurpa D. ThungonDocument15 pagesFood & Genes: Term Paper By: Phurpa D. ThungonMehul BhardwajNo ratings yet
- 1 s2.0 S092544391300327X MainDocument8 pages1 s2.0 S092544391300327X Main邢振凯No ratings yet
- Ijms 16 17193Document38 pagesIjms 16 17193Afaq AhmadNo ratings yet
- Oxidative Phosporylation in Cancer CellsDocument9 pagesOxidative Phosporylation in Cancer CellsM Naufal IlmiNo ratings yet
- Pharmacological Research: Nabila Bourebaba, Krzysztof MaryczDocument10 pagesPharmacological Research: Nabila Bourebaba, Krzysztof Marycz2m96k96vp8No ratings yet
- Diseases ArticleDocument15 pagesDiseases ArticlefreedogssmNo ratings yet
- Mitochondrial Correction A New Therapeutic Paradigm For Cancer and Degenerative Diseases JOM 33.4Document20 pagesMitochondrial Correction A New Therapeutic Paradigm For Cancer and Degenerative Diseases JOM 33.4La Isla OesteNo ratings yet
- 1-C Metabolism-Serine, Glycine, Folates - in Acute Myeloid LeukemiaDocument10 pages1-C Metabolism-Serine, Glycine, Folates - in Acute Myeloid LeukemiaVitoria LimaNo ratings yet
- Chaperoane 2005 Cystic FibrosisDocument11 pagesChaperoane 2005 Cystic FibrosisLiviu Athos TamasNo ratings yet
- Metab Energ de La Caquexia 2019Document13 pagesMetab Energ de La Caquexia 2019NaniihSotomayorNo ratings yet
- NIH Public Access: Author ManuscriptDocument5 pagesNIH Public Access: Author ManuscriptDayane CruzNo ratings yet
- The Oncologist 2004 Vaupel 10 7Document8 pagesThe Oncologist 2004 Vaupel 10 7Trịnh Vạn NgữNo ratings yet
- Kit Ada 2021Document15 pagesKit Ada 2021Gemeneza GonzalesNo ratings yet
- Au Top HagiaDocument48 pagesAu Top HagiaJorge CamargoNo ratings yet
- Aysegül Zeyno Epigenetic and Nutritient Lecture - New SunumDocument26 pagesAysegül Zeyno Epigenetic and Nutritient Lecture - New SunumzeynoleeeNo ratings yet
- News & Views: Surviving Import FailureDocument2 pagesNews & Views: Surviving Import FailureatharvatanksaleNo ratings yet
- Cancer MetabolismDocument10 pagesCancer MetabolismPrasad PhapaleNo ratings yet
- Paper 2 PDFDocument36 pagesPaper 2 PDFOmar Arias AndradeNo ratings yet
- Lucock 2011 Folic Acid Beyond MetabolismDocument12 pagesLucock 2011 Folic Acid Beyond Metabolismadinda.larastitiNo ratings yet
- Lipid Signaling and Lipotoxicity in Metaflammation Indications For Metabolic Disease Pathogenesis and TreatmentDocument16 pagesLipid Signaling and Lipotoxicity in Metaflammation Indications For Metabolic Disease Pathogenesis and TreatmentSherin KhodierNo ratings yet
- Cancer Metabolism in Space and Time - Beyond The Warburg EffectDocument17 pagesCancer Metabolism in Space and Time - Beyond The Warburg Effectyylf2016No ratings yet
- Intl Journal of Cancer - 2004 - Andersson - Downregulation of The Antiapoptotic MCL 1 Protein and Apoptosis in MA 11 BreastDocument9 pagesIntl Journal of Cancer - 2004 - Andersson - Downregulation of The Antiapoptotic MCL 1 Protein and Apoptosis in MA 11 BreastWojciech WawrętyNo ratings yet
- Chaperone Machines For Protein Folding, Unfolding and DisaggregationDocument13 pagesChaperone Machines For Protein Folding, Unfolding and DisaggregationPierre ChaulierNo ratings yet
- Role of ROS in Metabolic Diseases and ChronicDocument9 pagesRole of ROS in Metabolic Diseases and ChronicFian AldyNo ratings yet
- Neurodegenerative Diseases Multifactorial ConformationalDocument9 pagesNeurodegenerative Diseases Multifactorial ConformationalLala FemNo ratings yet
- Cheat Sheet (Bio) 1Document1 pageCheat Sheet (Bio) 1Kelvin KohNo ratings yet
- Review Article Autophagy in Acute Pancreatitis: Organelle Interaction and Microrna RegulationDocument12 pagesReview Article Autophagy in Acute Pancreatitis: Organelle Interaction and Microrna RegulationCésar CarvalloNo ratings yet
- Late-Onset Alzheimers Disease Is Associated WithDocument13 pagesLate-Onset Alzheimers Disease Is Associated WithAlix AliNo ratings yet
- Lab Invest 2014131 ADocument12 pagesLab Invest 2014131 AGeorge Sebastian AntonyNo ratings yet
- Ars 2009 2816Document32 pagesArs 2009 2816Nilabh RanjanNo ratings yet
- Drug ToxicityDocument21 pagesDrug ToxicityUsman Akhtar0% (1)
- The Hallmarks of CancerDocument6 pagesThe Hallmarks of CancerSaffronMaeNo ratings yet
- Agriculture 11 00407Document12 pagesAgriculture 11 00407EMEKA IKECHUKWUNo ratings yet
- Growing Beetroot SuccessfullyDocument5 pagesGrowing Beetroot SuccessfullyEMEKA IKECHUKWUNo ratings yet
- Eggplant Production Under Deficit Irrigation and Polyethylene MulchDocument15 pagesEggplant Production Under Deficit Irrigation and Polyethylene MulchEMEKA IKECHUKWUNo ratings yet
- Ria 1Document3 pagesRia 1EMEKA IKECHUKWUNo ratings yet
- Miracle IT Report by Emeka Ikechukwu EzekielDocument50 pagesMiracle IT Report by Emeka Ikechukwu EzekielEMEKA IKECHUKWUNo ratings yet
- Emeka Ikechukwu Power PointDocument16 pagesEmeka Ikechukwu Power PointEMEKA IKECHUKWUNo ratings yet
- UntitledDocument29 pagesUntitledEMEKA IKECHUKWUNo ratings yet
- IT Report by Emeka Ikechukwu EzekielDocument3 pagesIT Report by Emeka Ikechukwu EzekielEMEKA IKECHUKWUNo ratings yet
- Bacteria BioluminescenceDocument32 pagesBacteria BioluminescenceEMEKA IKECHUKWUNo ratings yet
- Check Unit 556 December Digestive PDFDocument33 pagesCheck Unit 556 December Digestive PDFdragon66No ratings yet
- Hemmingsen Et Al-2017-Cochrane Database of Systematic ReviewsDocument192 pagesHemmingsen Et Al-2017-Cochrane Database of Systematic ReviewsUfiq MantoNo ratings yet
- ImciDocument27 pagesImciRosalyn Marie Morales SugayNo ratings yet
- Macular Hole - EyeWikiDocument4 pagesMacular Hole - EyeWikiRaesoo90210No ratings yet
- NR 325 Diabetes Power-Student Copy1118Document39 pagesNR 325 Diabetes Power-Student Copy1118John MixerNo ratings yet
- Word Jurnal THT Odontogenik 1Document20 pagesWord Jurnal THT Odontogenik 1RayNo ratings yet
- Take These Steps To Avoid Allergens and Other Asthma Triggers That Can Interfere With Your SleepDocument8 pagesTake These Steps To Avoid Allergens and Other Asthma Triggers That Can Interfere With Your Sleeps.s.r.k.m. guptaNo ratings yet
- Careplan Week 8Document3 pagesCareplan Week 8api-353681121No ratings yet
- Drug StudyDocument4 pagesDrug StudyCyrene Jasmine CabañesNo ratings yet
- Dapoxetine Dosing InformationDocument25 pagesDapoxetine Dosing InformationA.R.AthreyaNo ratings yet
- RubellaDocument49 pagesRubellarusdiNo ratings yet
- Intensive Blood-Pressure Control in Hypertensive Chronic Kidney DiseaseDocument24 pagesIntensive Blood-Pressure Control in Hypertensive Chronic Kidney DiseaseMeldy Muzada ElfaNo ratings yet
- Chicken Pox HandoutsDocument4 pagesChicken Pox Handoutsmaglangitjoannamarie1920No ratings yet
- Differential DiagnosisDocument10 pagesDifferential DiagnosisRhaffy Bearneza RapaconNo ratings yet
- 7 Nefropatia Gravidica EnglDocument29 pages7 Nefropatia Gravidica EnglMadalina SercaianuNo ratings yet
- #4 - NCM 109 - TransesDocument22 pages#4 - NCM 109 - TransesJaimie BanaagNo ratings yet
- Long-Term Outcome in Congenitally Corrected Transposition of The Great ArteriesDocument7 pagesLong-Term Outcome in Congenitally Corrected Transposition of The Great Arteriespaul00040No ratings yet
- Anatomi Dan Fisiologi Mata 1Document61 pagesAnatomi Dan Fisiologi Mata 1MAWANNo ratings yet
- HemoglobinopatiDocument40 pagesHemoglobinopatiHenni Junita Siregar SorminNo ratings yet
- Early Postoperative Small Bowel Obstruction-The American Journal of Surgery 2015Document6 pagesEarly Postoperative Small Bowel Obstruction-The American Journal of Surgery 2015Trí Cương NguyễnNo ratings yet
- Child Cerebral Pasly Guideline For CliniciansDocument2 pagesChild Cerebral Pasly Guideline For CliniciansAnonymous qbQIQaPlNo ratings yet
- Iii. Clinical Reasoning Questions-CollaborationDocument16 pagesIii. Clinical Reasoning Questions-CollaborationAjie ZamNo ratings yet
- Cephalosporins in Animals - DosagesDocument7 pagesCephalosporins in Animals - DosagesSunilNo ratings yet
- Measles Care Study-1Document28 pagesMeasles Care Study-1YourFav RNNo ratings yet
- HIV and AIDSDocument8 pagesHIV and AIDSmariam miladNo ratings yet
- Hemorrhoids FinalDocument95 pagesHemorrhoids FinalregysujitNo ratings yet
- Luliconazole, A New Antifungal Against Candida Species Isolated From Different SourcesDocument5 pagesLuliconazole, A New Antifungal Against Candida Species Isolated From Different SourcesLina LimNo ratings yet
- Att 1446693658204 المذكرة-الذهبيةDocument80 pagesAtt 1446693658204 المذكرة-الذهبيةShady KhamisNo ratings yet
- Academiclinc OFA PPT V6 Jan 2022 (English) OriginalDocument282 pagesAcademiclinc OFA PPT V6 Jan 2022 (English) OriginalPARIMALA A/P NAGAN MoeNo ratings yet