Download as pdf or txt
You might also like
- Project-Proposal (Technopreneurship)Document18 pagesProject-Proposal (Technopreneurship)Luke MoraledaNo ratings yet
- Development of Non Dairy Whipping Cream PDFDocument3 pagesDevelopment of Non Dairy Whipping Cream PDFgutierrez_rgcNo ratings yet
- Comparison of The Physicochemical Properties and Thermal StabilityDocument13 pagesComparison of The Physicochemical Properties and Thermal StabilityLucas SchindelNo ratings yet
- Biochar ThermocatalyticDocument11 pagesBiochar ThermocatalyticGRagaNo ratings yet
- Recent Advances in Enzymatic Conversion of Lignin To Value Added ProductsDocument33 pagesRecent Advances in Enzymatic Conversion of Lignin To Value Added ProductsDark-ObsessionsNo ratings yet
- Inhibition of Ethanol Producing Yeast AnDocument17 pagesInhibition of Ethanol Producing Yeast Anveronica RodriguezNo ratings yet
- Lignin Method For VanillinDocument7 pagesLignin Method For VanillinShrutiNo ratings yet
- 2016 - Heterogeneously Catalyzed Lignin DepolymerizationDocument14 pages2016 - Heterogeneously Catalyzed Lignin DepolymerizationSHREE RAVVANo ratings yet
- 1 s2.0 S0926669015304842 MainDocument6 pages1 s2.0 S0926669015304842 Mainjarek3aNo ratings yet
- BiorrefinariaDocument39 pagesBiorrefinariaCândido LelisNo ratings yet
- Olsson 1996Document20 pagesOlsson 1996Jean Escobar CNo ratings yet
- Highly Efficient Liquefaction of Woody Biomass in Hot-Compressed Alcohol - Water Co-SolventsDocument9 pagesHighly Efficient Liquefaction of Woody Biomass in Hot-Compressed Alcohol - Water Co-SolventsBakir JusicNo ratings yet
- 10.1016 j.cogsc.2019.11.004-LECTURA 2-CLASE 4Document19 pages10.1016 j.cogsc.2019.11.004-LECTURA 2-CLASE 4ladyNo ratings yet
- 1 s2.0 S0360544213003800 MainDocument9 pages1 s2.0 S0360544213003800 MainPravaNo ratings yet
- The Direct Pyrolysis and Catalytic Pyrolysis of Nannochloropsis Sp. Residue For Renewable Bio-OilsDocument8 pagesThe Direct Pyrolysis and Catalytic Pyrolysis of Nannochloropsis Sp. Residue For Renewable Bio-OilsYohn Palomino BellidoNo ratings yet
- Bioresource Technology: Ke Zhang, Zhijian Pei, Donghai WangDocument13 pagesBioresource Technology: Ke Zhang, Zhijian Pei, Donghai WangAna Sofia Rojas CarpioNo ratings yet
- Mechanical Properties and Water Absorption of Glass Fibre Reinforced Bio-Phenolic Elastomer (BPE) CompositeDocument6 pagesMechanical Properties and Water Absorption of Glass Fibre Reinforced Bio-Phenolic Elastomer (BPE) Compositemalkinada179No ratings yet
- SSRN Id4313016Document18 pagesSSRN Id4313016Khelil OmarNo ratings yet
- BioRes 06 3 2647 ElMansouri YH Char Alkaline Lignins PF Epoxy Resins 1557Document16 pagesBioRes 06 3 2647 ElMansouri YH Char Alkaline Lignins PF Epoxy Resins 1557039 Asma SanjumNo ratings yet
- 1 s2.0 S0141813021023680 MainDocument9 pages1 s2.0 S0141813021023680 Mainmartina.cirronis96No ratings yet
- Lignin - From Natural Adsorbent To Activated Carbon A ReviewDocument12 pagesLignin - From Natural Adsorbent To Activated Carbon A ReviewyemresimsekNo ratings yet
- Hadad Et Al-2005-Journal of Applied MicrobiologyDocument8 pagesHadad Et Al-2005-Journal of Applied MicrobiologynahrawiNo ratings yet
- Extraction of Phenols From Lignin Microwave-Pyrolysis Oil Using A Switchable Hydrophilicity Solvent PDFDocument8 pagesExtraction of Phenols From Lignin Microwave-Pyrolysis Oil Using A Switchable Hydrophilicity Solvent PDFjiiNo ratings yet
- J Ijbiomac 2021 07 082Document9 pagesJ Ijbiomac 2021 07 082Douglas SantosNo ratings yet
- TMP 1 DE5Document8 pagesTMP 1 DE5FrontiersNo ratings yet
- C8GC00502HDocument19 pagesC8GC00502HGRagaNo ratings yet
- Lignin 1Document13 pagesLignin 1nausheneNo ratings yet
- Laccase Mediator Pretreatment of Wheat StrawDocument15 pagesLaccase Mediator Pretreatment of Wheat StrawMeyerling RandfordNo ratings yet
- Conversion of Lignin To Aromatic-Based Chemicals (L-Chems) and Biofuels (L-Fuels)Document7 pagesConversion of Lignin To Aromatic-Based Chemicals (L-Chems) and Biofuels (L-Fuels)Claudia Elizabeth Ruiz DávilaNo ratings yet
- Research Article: Enhanced Ethanol and Biogas Production From Pinewood by NMMO Pretreatment and Detailed Biomass AnalysisDocument11 pagesResearch Article: Enhanced Ethanol and Biogas Production From Pinewood by NMMO Pretreatment and Detailed Biomass AnalysislailaNo ratings yet
- Bioresource Technology: Kai Wörmeyer, Thomas Ingram, Bodo Saake, Gerd Brunner, Irina SmirnovaDocument8 pagesBioresource Technology: Kai Wörmeyer, Thomas Ingram, Bodo Saake, Gerd Brunner, Irina Smirnovadhy182No ratings yet
- Punya Rizky 1Document12 pagesPunya Rizky 1Chindy FebriNo ratings yet
- Hydrothermal Conversion of Lignin To Substituted Phenols and AromaticDocument4 pagesHydrothermal Conversion of Lignin To Substituted Phenols and AromaticClaudia Elizabeth Ruiz DávilaNo ratings yet
- Pretreatments To Enhance The Digestibility of Lignocellulosic BiomassDocument9 pagesPretreatments To Enhance The Digestibility of Lignocellulosic BiomassMarco Rezende100% (1)
- Bioresource TechnologyDocument7 pagesBioresource Technologykartik521No ratings yet
- Novel Bio-Degradable LIGNIN Reinforced NBR CompositesDocument16 pagesNovel Bio-Degradable LIGNIN Reinforced NBR CompositesjvchiqueNo ratings yet
- Accepted Manuscript: 10.1016/j.polymer.2017.02.036Document32 pagesAccepted Manuscript: 10.1016/j.polymer.2017.02.036Syafiq JaafarNo ratings yet
- Iz Aguirre 2020Document9 pagesIz Aguirre 2020Luciana BetzlerNo ratings yet
- 1 s2.0 S0141813023002209 MainDocument14 pages1 s2.0 S0141813023002209 MainÖykü DemirelNo ratings yet
- International Biodeterioration & Biodegradation: Miriam Santo, Ronen Weitsman, Alex SivanDocument7 pagesInternational Biodeterioration & Biodegradation: Miriam Santo, Ronen Weitsman, Alex SivanZero HeroNo ratings yet
- 1 SD PDFDocument10 pages1 SD PDFDaniel Salazar DiazNo ratings yet
- DerivativesApplicationsofLignin AnInsightDocument7 pagesDerivativesApplicationsofLignin AnInsightMaria Angelica Bacay BisaNo ratings yet
- 1) Bio-Based Epoxy Resins From Biorefinery By-Products PDFDocument10 pages1) Bio-Based Epoxy Resins From Biorefinery By-Products PDFkevin steve velandia avilaNo ratings yet
- 1 s2.0 S014374961730009X MainDocument8 pages1 s2.0 S014374961730009X MainDavid Fernández osorioNo ratings yet
- Industrial Crops & ProductsDocument11 pagesIndustrial Crops & ProductsAleksandrs ArnautovsNo ratings yet
- Boundzanga 2020Document16 pagesBoundzanga 2020Ingrid ContrerasNo ratings yet
- TLC Plate of BHETDocument6 pagesTLC Plate of BHETbitisa5368No ratings yet
- Lipase-Catalyzed Synthesis and Characterization of Biodegradable Polyester Containing - Malic Acid Unit in Solvent SystemDocument9 pagesLipase-Catalyzed Synthesis and Characterization of Biodegradable Polyester Containing - Malic Acid Unit in Solvent SystemIulia Georgiana SosoiNo ratings yet
- AlakliiDocument9 pagesAlakliiJuan RodriguezNo ratings yet
- Organic Fertilizer Soil SC 102Document40 pagesOrganic Fertilizer Soil SC 102Ruellyn Dura DumajelNo ratings yet
- Gregorova Et Al-2007-Journal of Applied Polymer Science PDFDocument6 pagesGregorova Et Al-2007-Journal of Applied Polymer Science PDFLaura Jane PereiraNo ratings yet
- The Role of Pretreatment in Improving The Enzymatic Hydrolysis of Lignocellulosic MaterialsDocument10 pagesThe Role of Pretreatment in Improving The Enzymatic Hydrolysis of Lignocellulosic MaterialsdazylahNo ratings yet
- Journal of Hazardous Materials: Jiajin Liang, Xiuxiu Fang, Yunqin Lin, Dehan WangDocument8 pagesJournal of Hazardous Materials: Jiajin Liang, Xiuxiu Fang, Yunqin Lin, Dehan WangJayNo ratings yet
- Bioresource Technology: Mailin Misson, Roslindawati Haron, Mohd Fadhzir Ahmad Kamaroddin, Nor Aishah Saidina AminDocument7 pagesBioresource Technology: Mailin Misson, Roslindawati Haron, Mohd Fadhzir Ahmad Kamaroddin, Nor Aishah Saidina Aminwidianingsih100% (1)
- Influence of Steaming Pressure On Steam Explosion Pretreatment of Lespedeza Stalks (Lespedeza Cyrtobotrya) - II. Characteristics of Degraded LigninDocument9 pagesInfluence of Steaming Pressure On Steam Explosion Pretreatment of Lespedeza Stalks (Lespedeza Cyrtobotrya) - II. Characteristics of Degraded Ligninnuljmal_535260430No ratings yet
- Mattsson 2016Document14 pagesMattsson 2016John ZoidbergNo ratings yet
- Minami DKK., 2003Document9 pagesMinami DKK., 2003inershit studioNo ratings yet
- Pengaruh Perlakuan Delignifikasi Terhadap Hidrolisis Selulosa Dan Produksi Etanol Dari Limbah BerlignoselulosaDocument12 pagesPengaruh Perlakuan Delignifikasi Terhadap Hidrolisis Selulosa Dan Produksi Etanol Dari Limbah BerlignoselulosaRiana KhoirunnisaNo ratings yet
- Separation and Puri Fication Technology: SciencedirectDocument7 pagesSeparation and Puri Fication Technology: SciencedirectMariaNo ratings yet
- Lignin, Lignocellulose, LigninaseDocument12 pagesLignin, Lignocellulose, LigninasectnbsNo ratings yet
- Lignin Removal and Benzene-Alcohol Extraction Effects On Lignin Measurements of The Hydrothermal Pretreated Bamboo Substrate PDFDocument5 pagesLignin Removal and Benzene-Alcohol Extraction Effects On Lignin Measurements of The Hydrothermal Pretreated Bamboo Substrate PDFjiiNo ratings yet
- Recent Advances in Polyphenol ResearchFrom EverandRecent Advances in Polyphenol ResearchHeidi HalbwirthNo ratings yet
- 1 s2.0 S0304389415000643 MainDocument13 pages1 s2.0 S0304389415000643 Mainla sourceNo ratings yet
- Chemical Data CollectionsDocument18 pagesChemical Data Collectionsla sourceNo ratings yet
- 1 s2.0 S2212371714000389 MainDocument14 pages1 s2.0 S2212371714000389 Mainla sourceNo ratings yet
- 1 s2.0 S0925963522003818 MainDocument10 pages1 s2.0 S0925963522003818 Mainla sourceNo ratings yet
- Optimization of Process Parameters For COD RemovalDocument6 pagesOptimization of Process Parameters For COD Removalla sourceNo ratings yet
- 1 s2.0 S0959652609002133 MainDocument5 pages1 s2.0 S0959652609002133 Mainla sourceNo ratings yet
- 1 s2.0 S0013468611006918 MainDocument6 pages1 s2.0 S0013468611006918 Mainla sourceNo ratings yet
- Li 2019 Recent Development of SupercapacitoDocument15 pagesLi 2019 Recent Development of Supercapacitola sourceNo ratings yet
- 1 s2.0 S0254058420304156 MainDocument10 pages1 s2.0 S0254058420304156 Mainla sourceNo ratings yet
- 1 s2.0 S2405844020302814 MainDocument10 pages1 s2.0 S2405844020302814 Mainla sourceNo ratings yet
- Forests 12 01160 v2Document19 pagesForests 12 01160 v2la sourceNo ratings yet
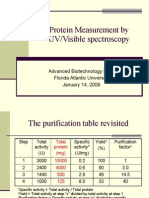
- Protein Measurement by UV/Visible Spectroscopy: Advanced Biotechnology Lab I Florida Atlantic University January 14, 2008Document10 pagesProtein Measurement by UV/Visible Spectroscopy: Advanced Biotechnology Lab I Florida Atlantic University January 14, 2008Gina ZhouNo ratings yet
- AFCONA - 4530 TDS EngDocument1 pageAFCONA - 4530 TDS EngMaleja HerediaNo ratings yet
- 7-Tontine Wall Ceiling BlanketDocument2 pages7-Tontine Wall Ceiling BlanketAdrian Law PangNo ratings yet
- How Does DCPIP WorkDocument3 pagesHow Does DCPIP WorkIsaac LeeNo ratings yet
- 763-Article Word Doc File-2585-1-10-20201201Document7 pages763-Article Word Doc File-2585-1-10-20201201HarshaNo ratings yet
- Carbonyl CompoundsDocument50 pagesCarbonyl CompoundsFariz SharudinNo ratings yet
- Haramaya University - Summer 2011Document4 pagesHaramaya University - Summer 2011aakuma100% (2)
- Distillation - Extractive Distillation: December 2013Document25 pagesDistillation - Extractive Distillation: December 2013حمامة السلامNo ratings yet
- Aromatic CompoundsDocument107 pagesAromatic CompoundsNishantNo ratings yet
- Alicyclic Chemistry - (2023)Document228 pagesAlicyclic Chemistry - (2023)sattar jabbar100% (2)
- Choosing A Well Format Contents and Storage: Quick Reference Pub. No. MAN0009818 Rev. C.0Document4 pagesChoosing A Well Format Contents and Storage: Quick Reference Pub. No. MAN0009818 Rev. C.0silmaril trehNo ratings yet
- Halophiles PDFDocument11 pagesHalophiles PDFWulan PurnamasariNo ratings yet
- CHE323 FS18 Teil1 - PDF PDFDocument114 pagesCHE323 FS18 Teil1 - PDF PDFreauhanNo ratings yet
- 1 s2.0 S1226086X2100410X MainDocument22 pages1 s2.0 S1226086X2100410X MainAndres Felipe Tamayo RodriguezNo ratings yet
- 1.3 JP2021195320A - TranslationDocument5 pages1.3 JP2021195320A - TranslationNilesh PatelNo ratings yet
- Unit 4 EqulibriaDocument2 pagesUnit 4 EqulibriaSahanNivanthaNo ratings yet
- # of Carbons Name Formula (C: AP Chemistry Chapter 22 - Organic ChemistryDocument5 pages# of Carbons Name Formula (C: AP Chemistry Chapter 22 - Organic ChemistryChemist Mohamed MohyNo ratings yet
- Summative Test BiomoleculesDocument1 pageSummative Test BiomoleculesMeryl GallardoNo ratings yet
- Product Manual 8 18072023151321 8Document10 pagesProduct Manual 8 18072023151321 8lakshya bagriNo ratings yet
- Lab SBL 3Document4 pagesLab SBL 3api-383478918No ratings yet
- YE 101 Lecture-5 JuteDocument17 pagesYE 101 Lecture-5 Jutejiban srNo ratings yet
- Ecoflex BrochureDocument24 pagesEcoflex BrochureRosman SenawiNo ratings yet
- Science Worksheet - SolutionDocument15 pagesScience Worksheet - SolutionZainul ShaikhNo ratings yet
- Art 24 Bio - Zhang2019 - ALGASDocument6 pagesArt 24 Bio - Zhang2019 - ALGASandrea acostaNo ratings yet
- Introduction To BiochemistryDocument5 pagesIntroduction To BiochemistryDr. Dhondiba VishwanathNo ratings yet
- Antioxidant and Anti-Inflammatory Studies of Leaf of Ricinus Communis and Rhizome of Curcuma Amada For Topical ApplicationDocument13 pagesAntioxidant and Anti-Inflammatory Studies of Leaf of Ricinus Communis and Rhizome of Curcuma Amada For Topical ApplicationInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- LESSON PLAN Isomer & Reaksi Alkana, Alkena & AlkunaDocument5 pagesLESSON PLAN Isomer & Reaksi Alkana, Alkena & AlkunaabinulNo ratings yet
- Extraction Adviser in enDocument8 pagesExtraction Adviser in enGhorpade GsmNo ratings yet