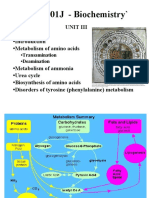
Download as docx, pdf, or txt
You might also like
- Lab Manual SCOA 20 21Document37 pagesLab Manual SCOA 20 21Anuj Padmawar0% (1)
- Environmental Science Your World Your Turn Chapter 5 NotesDocument32 pagesEnvironmental Science Your World Your Turn Chapter 5 NotesTaylorRayKane100% (2)
- 03 - Metabolisme Protein - MonogastrikDocument25 pages03 - Metabolisme Protein - MonogastrikHerni Bustam100% (1)
- Protein MetabolismDocument15 pagesProtein MetabolismAfshan Akhtar100% (1)
- Class Notes of Amino Acid MetabolismDocument49 pagesClass Notes of Amino Acid MetabolismShivanand Mali100% (1)
- Amino Acid Metabolism NotesDocument152 pagesAmino Acid Metabolism NotesAkhilesh TiwariNo ratings yet
- 6 Proteins Their Digestion & AbsorptionsDocument18 pages6 Proteins Their Digestion & AbsorptionshodaNo ratings yet
- Amino Acid Metabolism All LecturesDocument18 pagesAmino Acid Metabolism All Lecturesmizare29gNo ratings yet
- Topic: Protein Metabolism: Biochemistry & Biophysics: Paper-V, Unit-9Document14 pagesTopic: Protein Metabolism: Biochemistry & Biophysics: Paper-V, Unit-9aayushi tejwaniNo ratings yet
- 4 ProteinDocument11 pages4 ProteinconcepcionsofiakarylleNo ratings yet
- Non-Essential Amino Acids Nitrogen MetabolismDocument21 pagesNon-Essential Amino Acids Nitrogen Metabolismpradeep36No ratings yet
- Investigation of Plasma Protein DisordersDocument10 pagesInvestigation of Plasma Protein DisordersJosiah BimabamNo ratings yet
- 9a. Metabolisme Asam AminoDocument62 pages9a. Metabolisme Asam AminohimawarumNo ratings yet
- LifeScience I Week 2 Topic 3Document36 pagesLifeScience I Week 2 Topic 3nsemn1234No ratings yet
- CHAPTER 4 - BodyBuilding Foods - ProteinDocument8 pagesCHAPTER 4 - BodyBuilding Foods - ProteinMary Jane TiangsonNo ratings yet
- Amino Acid MetabolismDocument15 pagesAmino Acid Metabolismshanto.tn98No ratings yet
- Amino AcidsDocument32 pagesAmino AcidsWakjira GemedaNo ratings yet
- Amino Acid Biosynthesis and DegradationDocument11 pagesAmino Acid Biosynthesis and DegradationFarhana AktharNo ratings yet
- 15BT103 Biochem-UNIT 3Document53 pages15BT103 Biochem-UNIT 3Adityanair RA1711009010128No ratings yet
- Amino Acid Peptides and ProteinsDocument21 pagesAmino Acid Peptides and ProteinsKuntal AdityaNo ratings yet
- Presented by "Prof. Dr. Masoom Khan" Lecture Notes. No. 678Document41 pagesPresented by "Prof. Dr. Masoom Khan" Lecture Notes. No. 678Qamar QamarNo ratings yet
- Protein MetabolismDocument23 pagesProtein MetabolismSafura IjazNo ratings yet
- Amino Acid Catabolism II: Dr. Mohammad AkramDocument19 pagesAmino Acid Catabolism II: Dr. Mohammad Akramapi-19824406No ratings yet
- Biochemistry Unit 03 PDFDocument66 pagesBiochemistry Unit 03 PDFDhanush kannanNo ratings yet
- S1 Bioqui RenalDocument46 pagesS1 Bioqui RenalIsabela Cristina Verdugo SinibaldiNo ratings yet
- Tips 11 20Document43 pagesTips 11 20ChristianNo ratings yet
- A Comparative Study of Protein Content in Eggs and Soy ChunksDocument33 pagesA Comparative Study of Protein Content in Eggs and Soy ChunksNamratha DevadigaNo ratings yet
- Chemical Pathology BS-MLT 5Th SemesterDocument36 pagesChemical Pathology BS-MLT 5Th SemesterMuhammad AbdullahNo ratings yet
- Amino Acid MetabolismDocument10 pagesAmino Acid MetabolismatifzeaNo ratings yet
- Lecture 4 Protein Metabolism 1Document18 pagesLecture 4 Protein Metabolism 1beneficialboxer9237No ratings yet
- Protein & Amino Acid MetabolismDocument196 pagesProtein & Amino Acid MetabolismSantino MajokNo ratings yet
- Biochemistery:: Asparagine Is The 1Document12 pagesBiochemistery:: Asparagine Is The 1Naima AmjadNo ratings yet
- AMINO ACIDS (Edited)Document126 pagesAMINO ACIDS (Edited)Keez HaruNo ratings yet
- Essential Amino AcidsDocument3 pagesEssential Amino AcidsDrbee10No ratings yet
- Protein and Amino Acid MetabolismDocument52 pagesProtein and Amino Acid MetabolismRisky OpponentNo ratings yet
- Proteins CC1Document6 pagesProteins CC1Aysha AishaNo ratings yet
- Amino Acids Metabol Synth of UreaDocument32 pagesAmino Acids Metabol Synth of UreaAnastasiafynn100% (1)
- Amino Acids Metabolism Final For Pharm 2014Document57 pagesAmino Acids Metabolism Final For Pharm 2014Getu LuchesaNo ratings yet
- Amino AcidsDocument30 pagesAmino AcidsroymalindoNo ratings yet
- BIOSINTESIS PROTlanjutanDocument31 pagesBIOSINTESIS PROTlanjutanAprilikkaearlyNo ratings yet
- Protein, Nitrogen Katabolisme Dan Siklus UreaDocument35 pagesProtein, Nitrogen Katabolisme Dan Siklus UreaAnonymous QCMhA4wNgBNo ratings yet
- C10 Protein and Amino Acid MetabolismDocument8 pagesC10 Protein and Amino Acid MetabolismSoraya D. Al-ObinayNo ratings yet
- Pathways in Amino Acids and Protein Metabolism 2Document76 pagesPathways in Amino Acids and Protein Metabolism 2godfreybwerere13No ratings yet
- Lect1 - 2017Document28 pagesLect1 - 2017George MakoriNo ratings yet
- Note On Digestion of Protein 2021Document11 pagesNote On Digestion of Protein 2021Charity NwaogbeNo ratings yet
- Investigation of Amino Acid Metabolism Disorders Series 1Document45 pagesInvestigation of Amino Acid Metabolism Disorders Series 1kiedd_04100% (4)
- Metabolism of Essential and Non-Essential Amino AcidsDocument15 pagesMetabolism of Essential and Non-Essential Amino AcidsMrs SalimNo ratings yet
- METABOLISME ASAM AMINO Protein BiologiDocument31 pagesMETABOLISME ASAM AMINO Protein BiologiAxzchiuu :vNo ratings yet
- Topic 3 Amino Acid and Protein-EditedDocument36 pagesTopic 3 Amino Acid and Protein-Editedشيكين LurveJellabiesNo ratings yet
- Protein BiochemistryDocument50 pagesProtein BiochemistrySudipta MandolNo ratings yet
- Metabolism of Essential AADocument7 pagesMetabolism of Essential AA48 Syeda KainatNo ratings yet
- Chapter 6 Chemistry of Proteins (Compatibility Mode)Document28 pagesChapter 6 Chemistry of Proteins (Compatibility Mode)bayan.grNo ratings yet
- Tips 11-20Document43 pagesTips 11-20Kenneth DayritNo ratings yet
- Nutrition Module 5Document7 pagesNutrition Module 5mimiNo ratings yet
- Proteins and Amino Acid-NoteDocument13 pagesProteins and Amino Acid-NoteHABIBUR RAHAMANNo ratings yet
- ProteinsDocument29 pagesProteinsOgwuche AndrewNo ratings yet
- Protein Metabolism..Document32 pagesProtein Metabolism..Ahmed JamalNo ratings yet
- What Is Amino AcidsDocument12 pagesWhat Is Amino AcidstollasbulldogsNo ratings yet
- Amino Acid For B.TechDocument4 pagesAmino Acid For B.TechAnushka AdhikariNo ratings yet
- Protein Digestion and AbsorptionDocument20 pagesProtein Digestion and AbsorptionMarvin JeaNo ratings yet
- Notes ProteinsDocument7 pagesNotes ProteinsJahanzeb SafdarNo ratings yet
- Approach To Chest PainDocument37 pagesApproach To Chest Painمصطفى عادل موسى احمدNo ratings yet
- Beta Oxidation of Saturated Fatty AcidsDocument15 pagesBeta Oxidation of Saturated Fatty Acidsمصطفى عادل موسى احمدNo ratings yet
- Presentation 3Document14 pagesPresentation 3مصطفى عادل موسى احمدNo ratings yet
- Community MedicineDocument3 pagesCommunity Medicineمصطفى عادل موسى احمدNo ratings yet
- School ReportDocument3 pagesSchool Reportمصطفى عادل موسى احمدNo ratings yet
- Chapter 11Document30 pagesChapter 11popayonutz22No ratings yet
- Avida ED ExercisesDocument44 pagesAvida ED ExercisesLara Patricia Causo0% (1)
- Kgmu Thesis 19.06 PDFDocument139 pagesKgmu Thesis 19.06 PDFavnish100% (1)
- Factors Affecting Starch DigestionDocument2 pagesFactors Affecting Starch DigestionNikki Ticman100% (1)
- Understanding OsteoporosisDocument6 pagesUnderstanding OsteoporosisPriyank GuptaNo ratings yet
- Microwave Extraction of Polyphenol From Pomegranate Seed: Original Research ArticleDocument11 pagesMicrowave Extraction of Polyphenol From Pomegranate Seed: Original Research ArticleFaycel OuerdienNo ratings yet
- Toma de Decisiones: en Adolescentes y Autoconfianza:, Ana Raquel Gutierrez CDocument6 pagesToma de Decisiones: en Adolescentes y Autoconfianza:, Ana Raquel Gutierrez CJhuvenal UmiyauriNo ratings yet
- 02 - Control and CoordinationDocument20 pages02 - Control and CoordinationSamveg ClassesNo ratings yet
- Specimen Collection and ProcessingDocument7 pagesSpecimen Collection and ProcessingJangHanbyul100% (1)
- MRI of Enthesitis in As (Including PsA)Document7 pagesMRI of Enthesitis in As (Including PsA)wandering_bearNo ratings yet
- Mamals of Kerala PDFDocument164 pagesMamals of Kerala PDFAnand SundaramNo ratings yet
- Issues and Challenges of Land Policy in Malaysia Relate To Customary Land and Malay Reserve LandDocument11 pagesIssues and Challenges of Land Policy in Malaysia Relate To Customary Land and Malay Reserve LandAizuddinHakimBenny0% (1)
- (Methods in Molecular Biology 1257) Willem F. Wolkers, Harriëtte Oldenhof (Eds.) - Cryopreservation and Freeze-Drying Protocols-Springer-Verlag New York (2015)Document505 pages(Methods in Molecular Biology 1257) Willem F. Wolkers, Harriëtte Oldenhof (Eds.) - Cryopreservation and Freeze-Drying Protocols-Springer-Verlag New York (2015)Sebastián Lescano100% (1)
- 6.1.180.heparin Sodium (Heparinum Natricum)Document2 pages6.1.180.heparin Sodium (Heparinum Natricum)Marcelino NoviantoNo ratings yet
- Bio 110 Introduction To Biology and Society Fall 2019 Course SyllabusDocument10 pagesBio 110 Introduction To Biology and Society Fall 2019 Course SyllabusJohn MixerNo ratings yet
- Plant Viruses ThesisDocument7 pagesPlant Viruses ThesisHelpInWritingPaperCanada100% (2)
- Sih Et Al. 2004. Behavioural Syndromes. An Ecological and Evolutionary OverviewDocument7 pagesSih Et Al. 2004. Behavioural Syndromes. An Ecological and Evolutionary OverviewKathiNo ratings yet
- Application of PCR-Based Methods To Assess The InfDocument12 pagesApplication of PCR-Based Methods To Assess The InfLucas Emanuel Freitas PereiraNo ratings yet
- Anti-Cancer Drugs Sir WaseemDocument29 pagesAnti-Cancer Drugs Sir WaseemglobalaffairzNo ratings yet
- Occult Meaning of Colours Purple Haze, All in My BrainDocument54 pagesOccult Meaning of Colours Purple Haze, All in My BrainrobinhoodlumNo ratings yet
- Apg 1Document13 pagesApg 1Manu MohamedNo ratings yet
- 2 Year - BS Medical Technology - 2 Sem - 1 BlockDocument20 pages2 Year - BS Medical Technology - 2 Sem - 1 BlockBelle Lat100% (1)
- Body Systems Lesson PlanDocument10 pagesBody Systems Lesson PlanadelNo ratings yet
- Feminism and Gender - Making Anthropology PublicDocument9 pagesFeminism and Gender - Making Anthropology PublicPearl SkyNo ratings yet
- Philippine National Standard: Halâl Agriculture and Fisheries ProductsDocument26 pagesPhilippine National Standard: Halâl Agriculture and Fisheries ProductsGaily HontiverosNo ratings yet
- Analytical, Toxicity, Experimental and Clinical Validation of Ayurvedic Formulations /herbal Crude DrugsDocument44 pagesAnalytical, Toxicity, Experimental and Clinical Validation of Ayurvedic Formulations /herbal Crude DrugsVasavi ChittemreddyNo ratings yet
- Blotting TechniquesDocument19 pagesBlotting TechniquesGhilli TecNo ratings yet
- Experiment No. 5 Starch Hydrolysis by AmylaseDocument10 pagesExperiment No. 5 Starch Hydrolysis by AmylasebobbymayaaNo ratings yet