Download as pdf or txt
You might also like
- Pub - Microbiology A Human Perspective PDFDocument884 pagesPub - Microbiology A Human Perspective PDFsamon sumulongNo ratings yet
- Cellular and Molecular PharmacologyFrom EverandCellular and Molecular PharmacologyRating: 4.5 out of 5 stars4.5/5 (6)
- Stam 2004Document12 pagesStam 2004CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Struct GenomDocument7 pagesStruct GenomNaidelyn HernandezNo ratings yet
- On The Accuracy of Homology Modeling and Sequence AlignmentDocument10 pagesOn The Accuracy of Homology Modeling and Sequence AlignmentCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Proteins: Remote Homology Detection of Integral Membrane Proteins Using Conserved Sequence FeaturesDocument13 pagesProteins: Remote Homology Detection of Integral Membrane Proteins Using Conserved Sequence FeaturesLavanya Priya SathyanNo ratings yet
- 2013 J Theor Biol 328 77-88Document12 pages2013 J Theor Biol 328 77-88mbrylinskiNo ratings yet
- Mustang PSFB FinalDocument49 pagesMustang PSFB FinalNiranjan BhuvanaratnamNo ratings yet
- Cavidad PrediccionDocument9 pagesCavidad PrediccionLuis MartinezNo ratings yet
- Pieper NucleicAcidsRes 2004Document6 pagesPieper NucleicAcidsRes 2004ctak2022No ratings yet
- Mol - Modelling of BCL-2 FinalDocument37 pagesMol - Modelling of BCL-2 FinalVannakumar SubramanianNo ratings yet
- COACH YZhangDocument8 pagesCOACH YZhangChristos FeidakisNo ratings yet
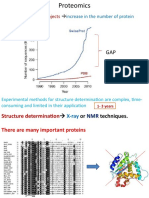
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- Homo LogyDocument8 pagesHomo LogyIgnacioEguinoaNo ratings yet
- 2011 J Struct Biol 173 558-569Document12 pages2011 J Struct Biol 173 558-569mbrylinskiNo ratings yet
- 2015 Article 14 Twilight ZoneDocument11 pages2015 Article 14 Twilight Zoneadiav PakNo ratings yet
- Homolgy ModelingDocument19 pagesHomolgy ModelingJainendra JainNo ratings yet
- Homology Modeling, Also Known As Comparative Modeling ofDocument19 pagesHomology Modeling, Also Known As Comparative Modeling ofManoharNo ratings yet
- Graph Based SignatureDocument8 pagesGraph Based SignatureNyasha ChitavatiNo ratings yet
- Bioinformatics Notes - 17Bt54: Module - 4Document48 pagesBioinformatics Notes - 17Bt54: Module - 4Samuel PrinceNo ratings yet
- Integrative Modeling of Membrane-Associated Protein AssembliesDocument12 pagesIntegrative Modeling of Membrane-Associated Protein AssembliesConstanza IsabellaNo ratings yet
- In Silico Prediction and Characterization of Protein Post Translational Modifications - 2016 - Journal of ProteomicsDocument11 pagesIn Silico Prediction and Characterization of Protein Post Translational Modifications - 2016 - Journal of ProteomicsmaikruspeNo ratings yet
- On Position-Specific Scoring Matrix For Protein Function PredictionDocument8 pagesOn Position-Specific Scoring Matrix For Protein Function PredictionW Antonio Muñoz ChNo ratings yet
- Transmembrane Protein Alignment and Fold Recognition Based On Predicted TopologyDocument9 pagesTransmembrane Protein Alignment and Fold Recognition Based On Predicted TopologyCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- 2010 Proteins 78 118-134Document17 pages2010 Proteins 78 118-134mbrylinskiNo ratings yet
- Protein Structure Prediction Using Homology ModelingDocument11 pagesProtein Structure Prediction Using Homology Modelingthelmamapangela64No ratings yet
- 10.IJAEST Vol No 7 Issue No 2 Human Protein Function Prediction An Overview 239 244Document6 pages10.IJAEST Vol No 7 Issue No 2 Human Protein Function Prediction An Overview 239 244helpdesk9532No ratings yet
- 2013 Front Genet 4 118Document7 pages2013 Front Genet 4 118mbrylinskiNo ratings yet
- TMP D73Document15 pagesTMP D73FrontiersNo ratings yet
- I-TASSER: A Unified Platform For Automated Protein Structure and Function PredictionDocument14 pagesI-TASSER: A Unified Platform For Automated Protein Structure and Function PredictionDavidNo ratings yet
- Comparison of The PAM and BLOSUM Amino Acid Substitution MatricesDocument4 pagesComparison of The PAM and BLOSUM Amino Acid Substitution Matricesmorteza hosseiniNo ratings yet
- 1 s2.0 S0969212611003364 MainDocument11 pages1 s2.0 S0969212611003364 Maintiago almeidaNo ratings yet
- 13 Application of Homology ModelingDocument7 pages13 Application of Homology Modelingklundermadona93No ratings yet
- Molecular Dynamics Simulations of Membrane Proteins: Philip C. Biggin and Peter J. BondDocument18 pagesMolecular Dynamics Simulations of Membrane Proteins: Philip C. Biggin and Peter J. BondKübra KahveciNo ratings yet
- Bertology Meets Biology: Interpreting Attention in Protein Language ModelsDocument22 pagesBertology Meets Biology: Interpreting Attention in Protein Language ModelsdisiskdsjNo ratings yet
- Bif501 AssDocument3 pagesBif501 Asssana 568aNo ratings yet
- Jurnal Alopatrik ENGDocument9 pagesJurnal Alopatrik ENGPuri RahmaNo ratings yet
- Dynamics RelateDocument11 pagesDynamics RelatevijayNo ratings yet
- Peerj Cs 275Document17 pagesPeerj Cs 275Binti SolihahNo ratings yet
- Efficient Protein Interaction Hotspot Identification in ProteinsDocument4 pagesEfficient Protein Interaction Hotspot Identification in ProteinsIJERDNo ratings yet
- Unit 1: Structural GenomicsDocument4 pagesUnit 1: Structural GenomicsLavanya ReddyNo ratings yet
- In Silico Attributes of Cold Resistance Protein 1 (Crp1) F Rom Brassica OleraceaeDocument8 pagesIn Silico Attributes of Cold Resistance Protein 1 (Crp1) F Rom Brassica OleraceaeresearchinventyNo ratings yet
- H ' I P P A: MM S Nterpolation of Rotiens For Rofile NalysisDocument10 pagesH ' I P P A: MM S Nterpolation of Rotiens For Rofile NalysisijcseitNo ratings yet
- Identification of Functionally Related Enzymes by Learning-to-Rank MethodsDocument13 pagesIdentification of Functionally Related Enzymes by Learning-to-Rank MethodsYahya HashmiNo ratings yet
- 2011 Rokas CPMBDocument14 pages2011 Rokas CPMBdina ratna komalaNo ratings yet
- Structural Category of Protein Membrane Protein: BBL741 Term Paper PresentationDocument7 pagesStructural Category of Protein Membrane Protein: BBL741 Term Paper PresentationMayank SinghNo ratings yet
- From Genomics To Proteomics: InsightDocument5 pagesFrom Genomics To Proteomics: Insightwadoud aggounNo ratings yet
- Homologymodeling 150123025144 Conversion Gate01Document27 pagesHomologymodeling 150123025144 Conversion Gate01Jhanvi SNo ratings yet
- Proteo MicsDocument25 pagesProteo MicsSaptarshi GhoshNo ratings yet
- Brylinski. FindsiteDocument6 pagesBrylinski. FindsiteRina HerowatiNo ratings yet
- Sanchez Bioinformatics 1999Document2 pagesSanchez Bioinformatics 1999hahahaNo ratings yet
- The Role of Protein Structure in Genomics: MinireviewDocument5 pagesThe Role of Protein Structure in Genomics: MinireviewAakriti SharmaNo ratings yet
- CPPS SandhyaDocument17 pagesCPPS SandhyaKenesei GyörgyNo ratings yet
- Bioinformatics 33-22-3549Document9 pagesBioinformatics 33-22-3549donkingdedon30No ratings yet
- High-Throughput Methods For Identification of Protein-Protein Interactions Involving Short Linear MotifsDocument9 pagesHigh-Throughput Methods For Identification of Protein-Protein Interactions Involving Short Linear Motifsraheleh mohammadi913No ratings yet
- Metabotools: A Comprehensive Toolbox For Analysis of Genome-Scale Metabolic ModelsDocument24 pagesMetabotools: A Comprehensive Toolbox For Analysis of Genome-Scale Metabolic ModelsJoão MoreiraNo ratings yet
- Image-Based Spatiotemporal Causality Inference For Protein Signaling NetworksDocument8 pagesImage-Based Spatiotemporal Causality Inference For Protein Signaling Networksmtallis3142275No ratings yet
- 2007 Int J Bioinform Res Appl 3 234-260Document27 pages2007 Int J Bioinform Res Appl 3 234-260mbrylinskiNo ratings yet
- Basics of ProteomicsDocument7 pagesBasics of ProteomicskonndaaiahNo ratings yet
- Functional Analysis of Genes: DOI: 10.2478/v10052-010-0001-YDocument16 pagesFunctional Analysis of Genes: DOI: 10.2478/v10052-010-0001-YTriều Nguyễn Phan ThúyNo ratings yet
- Bioinformation Discovery: Data to Knowledge in BiologyFrom EverandBioinformation Discovery: Data to Knowledge in BiologyNo ratings yet
- 0 AlignMeDocument6 pages0 AlignMeCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- New Alignment Strategy For TransmembraneDocument9 pagesNew Alignment Strategy For TransmembraneCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Diapositiva 2012tm-Coffee-130709120840-Phpapp02Document24 pagesDiapositiva 2012tm-Coffee-130709120840-Phpapp02CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- BALIBASE7REFDocument4 pagesBALIBASE7REFCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- On The Accuracy of Homology Modeling and Sequence AlignmentDocument10 pagesOn The Accuracy of Homology Modeling and Sequence AlignmentCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Stam 2004Document12 pagesStam 2004CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Bioinformatics: PHAT: A Transmembrane-Specific Substitution MatrixDocument7 pagesBioinformatics: PHAT: A Transmembrane-Specific Substitution MatrixCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Praline 3Document18 pagesPraline 3CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Survey, Comparison and Evaluation of Cross Platform Mobile Application Development ToolsDocument6 pagesSurvey, Comparison and Evaluation of Cross Platform Mobile Application Development ToolsCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Transmembrane Protein Alignment and Fold Recognition Based On Predicted TopologyDocument9 pagesTransmembrane Protein Alignment and Fold Recognition Based On Predicted TopologyCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- SQLLite Database and ListView AdapterDocument19 pagesSQLLite Database and ListView AdapterCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- PSITM-Coffee A Web SERVERDocument5 pagesPSITM-Coffee A Web SERVERCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Mobile First As A Best Practice in Web Design: Otto VarrelaDocument41 pagesMobile First As A Best Practice in Web Design: Otto VarrelaCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Ekstraksi, Purifikasi Dan Prinsip Kloning Dna: Agustina Setiawati, M.SC., AptDocument65 pagesEkstraksi, Purifikasi Dan Prinsip Kloning Dna: Agustina Setiawati, M.SC., AptDessy LipiNo ratings yet
- 0610 m16 Ms 42Document10 pages0610 m16 Ms 42Keshav JindalNo ratings yet
- EarthLifeSci PPT Q2W1Document24 pagesEarthLifeSci PPT Q2W1arvin paruliNo ratings yet
- MATH3353 NotesDocument100 pagesMATH3353 NotesOwen CameronNo ratings yet
- CAPE Biology 2011 U2 P2 MS Missing Page 14Document15 pagesCAPE Biology 2011 U2 P2 MS Missing Page 14Yagna LallNo ratings yet
- Teknik ARMS PCRDocument4 pagesTeknik ARMS PCRliyanaNo ratings yet
- Molecular Biology Sars-Cov-2 (Covid-19) Detection by Qualitative RT-PCRDocument2 pagesMolecular Biology Sars-Cov-2 (Covid-19) Detection by Qualitative RT-PCRAaryan K MNo ratings yet
- Chemical Mediators of Acute Inflammation 2Document41 pagesChemical Mediators of Acute Inflammation 2Sashika Tharindu100% (1)
- What Is A Cell - MedlinePlus GeneticsDocument3 pagesWhat Is A Cell - MedlinePlus Geneticswaqar ahmadNo ratings yet
- S.Y.B.Sc. Microbiology (MB-211 - 212 - 221 - 222) Question Bank PDFDocument33 pagesS.Y.B.Sc. Microbiology (MB-211 - 212 - 221 - 222) Question Bank PDFVivek MishraNo ratings yet
- Endocrinology Part 2Document6 pagesEndocrinology Part 2Prabhjot MundiNo ratings yet
- Leaf Quality AssessmentDocument3 pagesLeaf Quality Assessmentwaziri maulidiNo ratings yet
- Transcriptional Control of Floral Development: © 2017 American Society of Plant BiologistsDocument24 pagesTranscriptional Control of Floral Development: © 2017 American Society of Plant BiologistsCostinela SamoilăNo ratings yet
- Chapter3 FluorescenceDocument49 pagesChapter3 FluorescenceGuillaumeNo ratings yet
- Calculating Macronutrients EbookDocument36 pagesCalculating Macronutrients EbookPiyush100% (5)
- Berry Full of DNADocument15 pagesBerry Full of DNA3leniNo ratings yet
- PCR LectureDocument35 pagesPCR LectureArfan Tri KusumaNo ratings yet
- Chapter 13 Genetics and BiotechnologyDocument43 pagesChapter 13 Genetics and BiotechnologyMUHAMMAD SAALIM100% (2)
- SB 3Document8 pagesSB 3diyasanjeev13No ratings yet
- Fatty AcidsDocument31 pagesFatty Acidsdanena88No ratings yet
- Biochemistry: Questions & AnswersDocument9 pagesBiochemistry: Questions & AnswersMadani TawfeeqNo ratings yet
- The Genomics of The Farmed Shrimp Current Status and ApplicationDocument13 pagesThe Genomics of The Farmed Shrimp Current Status and ApplicationRama BangeraNo ratings yet
- HYbripol DNA PolymeraseDocument1 pageHYbripol DNA PolymeraseDanuNo ratings yet
- Learning Activity 2.3: Proteins: BiomoleculesDocument5 pagesLearning Activity 2.3: Proteins: BiomoleculesaikeeNo ratings yet
- Af9Pim500 0418M500 : Gotaq Hot Start PolymeraseDocument2 pagesAf9Pim500 0418M500 : Gotaq Hot Start Polymerasenbiolab6659No ratings yet
- Neurotransmission, Measuring Chemical Events In: Advanced ArticleDocument12 pagesNeurotransmission, Measuring Chemical Events In: Advanced ArticleazzaassNo ratings yet
- 5.3 Neuronal Communication POWERPOINTDocument58 pages5.3 Neuronal Communication POWERPOINTLisa MillardNo ratings yet
- Chromosomal Structure and Chromosomal MutationsDocument28 pagesChromosomal Structure and Chromosomal MutationsKavisa GhoshNo ratings yet
- Molecular Biology of The CellDocument129 pagesMolecular Biology of The CellAndres Sanchez EscobedoNo ratings yet