Download as docx, pdf, or txt
You might also like
- Pharmacoepidemiology, Pharmacoeconomics,PharmacovigilanceFrom EverandPharmacoepidemiology, Pharmacoeconomics,PharmacovigilanceRating: 3 out of 5 stars3/5 (1)
- AeromodellingDocument15 pagesAeromodellingzain ansariNo ratings yet
- (9789004316171 - Rashda - The Birth and Growth of An Egyptian Oasis Village) Rashda - The Birth and Growth of An Egyptian Oasis VillageDocument322 pages(9789004316171 - Rashda - The Birth and Growth of An Egyptian Oasis Village) Rashda - The Birth and Growth of An Egyptian Oasis VillageMarija RadjaNo ratings yet
- Drug Development Process: Preclinical StageDocument4 pagesDrug Development Process: Preclinical Stagepeter mwangiNo ratings yet
- Unit2 Clinical-TrialsDocument12 pagesUnit2 Clinical-TrialsDevangNo ratings yet
- An Introduction To Clinical TrialsDocument9 pagesAn Introduction To Clinical TrialsIrfan AhmadNo ratings yet
- 10 - Drug DevelopmentDocument5 pages10 - Drug DevelopmentLaura SaglietiNo ratings yet
- Clinical TrialDocument9 pagesClinical TrialAyanNo ratings yet
- Banned Drug 1Document6 pagesBanned Drug 1Nasrin Akther ShopnaNo ratings yet
- 2.2 Clinical Trails Phases - Clinical Research - Pharma DostDocument9 pages2.2 Clinical Trails Phases - Clinical Research - Pharma DostjhancyNo ratings yet
- The Drug Development ProcessDocument7 pagesThe Drug Development ProcessSACHIN BHASKAR NARKHEDE100% (1)
- Muhammad Umer (Assignment 01)Document6 pagesMuhammad Umer (Assignment 01)Umer KhanNo ratings yet
- Purpose of Drug DiscoveryDocument6 pagesPurpose of Drug DiscoveryUmer KhanNo ratings yet
- Different Phases I-IV of A Clinical Trial: Clinical Trials/Studies in HumansDocument5 pagesDifferent Phases I-IV of A Clinical Trial: Clinical Trials/Studies in HumansAshwan KumarNo ratings yet
- Exploring The Drug Development ProcessDocument9 pagesExploring The Drug Development Processs adhikariNo ratings yet
- Assignment On Drug DiscoveryDocument3 pagesAssignment On Drug DiscoveryMuhad KabeerNo ratings yet
- Drug DevelopmentDocument22 pagesDrug DevelopmentEAC School of Pharmacy ManilaNo ratings yet
- WWW - Clinicaltrials.gov: Clinical Phase I-IIDocument2 pagesWWW - Clinicaltrials.gov: Clinical Phase I-IIhelmanadyaNo ratings yet
- Clinical Trials: FDA ApprovalDocument3 pagesClinical Trials: FDA Approvalthamizh555No ratings yet
- GCP - Investigational New DrugsDocument14 pagesGCP - Investigational New DrugsRadio MiercolesNo ratings yet
- KeywordsDocument12 pagesKeywordsaadrika negiNo ratings yet
- Phases in Clinical TrialsDocument4 pagesPhases in Clinical TrialsManish Sarasvati100% (1)
- Investigational New DrugsDocument16 pagesInvestigational New DrugsVirgil CendanaNo ratings yet
- New Drug Application Content and Review Process For Clinical Pharmacology and BiopharmaceuticsDocument16 pagesNew Drug Application Content and Review Process For Clinical Pharmacology and BiopharmaceuticsAnki0391No ratings yet
- Pre MarketDocument12 pagesPre MarketNita Rezkiana AnwarNo ratings yet
- MPHR - 129 (Clinical Trial Managment)Document42 pagesMPHR - 129 (Clinical Trial Managment)Dr-Harikesh MauryaNo ratings yet
- 04 - The Drug Development Process - Step 3 - Clinical ResearchDocument5 pages04 - The Drug Development Process - Step 3 - Clinical ResearchArunendu MajiNo ratings yet
- Drug Development and ApprovalDocument16 pagesDrug Development and Approvalpulkit asatiNo ratings yet
- Types of Studies - NRC Research Institute (FDA)Document3 pagesTypes of Studies - NRC Research Institute (FDA)Muhammad ReyhanNo ratings yet
- Drug DevelopmentDocument5 pagesDrug DevelopmentJessica GlitterNo ratings yet
- Safety data generationDocument17 pagesSafety data generationAbhinav BhathejaNo ratings yet
- DRUG TESTING AND MEDICATION (By Anashe Magorimbo)Document2 pagesDRUG TESTING AND MEDICATION (By Anashe Magorimbo)Leon MzarabaniNo ratings yet
- Phases of Clinical TrialsDocument30 pagesPhases of Clinical TrialsMahum SohailNo ratings yet
- Presented By:-Bindu Xtreiy: Regn. No.: 12 Utdpr 0040Document51 pagesPresented By:-Bindu Xtreiy: Regn. No.: 12 Utdpr 0040parminder.nain29No ratings yet
- Clinical TrialsDocument73 pagesClinical TrialsSunilNo ratings yet
- 3 - 5 The Drug Development ProcessDocument10 pages3 - 5 The Drug Development Processaghanafissa5No ratings yet
- Step 3 - FDA Clinical Research - DrugsDocument6 pagesStep 3 - FDA Clinical Research - DrugsMuhammad ReyhanNo ratings yet
- Clinical Research: Presented By: Deepali KhetmalisDocument16 pagesClinical Research: Presented By: Deepali KhetmalisAmol KokaneNo ratings yet
- Clinical TrialsDocument21 pagesClinical TrialsvishakhaNo ratings yet
- Drug Development Process-1Document12 pagesDrug Development Process-1Ima AnNo ratings yet
- Practicals (Pharmaceutical Technology)Document24 pagesPracticals (Pharmaceutical Technology)Kustian Kohat100% (1)
- The Drug Development Process Step 3 - Clinical ResearchDocument5 pagesThe Drug Development Process Step 3 - Clinical ResearchDavid ThaiNo ratings yet
- Drug DevelopmentDocument5 pagesDrug Developmentnevelle4667No ratings yet
- Product Development and Technology Transfer Rushvi PatelDocument246 pagesProduct Development and Technology Transfer Rushvi Patelvidusha9727No ratings yet
- Regulatory Bodies in USA, Europe, India, China and AustraliaDocument9 pagesRegulatory Bodies in USA, Europe, India, China and AustraliapriyadarshNo ratings yet
- ANUBHAV DUBEY Phase of Clinical TrialsDocument4 pagesANUBHAV DUBEY Phase of Clinical Trialsgauravkumarsaras838No ratings yet
- Misha Regulatory AffairsDocument26 pagesMisha Regulatory AffairsGULSHAN MADHURNo ratings yet
- Peraturan Perudangan 2Document18 pagesPeraturan Perudangan 2YohanaNo ratings yet
- 9 - New Drug Development PlanDocument27 pages9 - New Drug Development PlanUmair MazharNo ratings yet
- Drug Development and EvaluationDocument25 pagesDrug Development and EvaluationArlises SinagaNo ratings yet
- Single-double-phaseIV-drug ApprovalDocument3 pagesSingle-double-phaseIV-drug ApprovalRoma Ann ManahanNo ratings yet
- Drug Development Process Cleveland, 6.23.06Document65 pagesDrug Development Process Cleveland, 6.23.06Srinivasa Chary SriramadasuNo ratings yet
- Phases of Clin TrialDocument26 pagesPhases of Clin TrialPraneeth Sanjeev ReddyNo ratings yet
- 2.3 Post Marketing Survaillence - Clinical Research - Pharma DostDocument7 pages2.3 Post Marketing Survaillence - Clinical Research - Pharma DostjhancyNo ratings yet
- Drug Discovery and DevelopmentDocument27 pagesDrug Discovery and Developmentshalini minocha100% (2)
- Ayush - GCP: DR Sathiya Rajeswaran Research Offi Cer SCRI, CCRS, ChennaiDocument34 pagesAyush - GCP: DR Sathiya Rajeswaran Research Offi Cer SCRI, CCRS, ChennaiGBKNo ratings yet
- Drug DVLPMT StgsDocument18 pagesDrug DVLPMT Stgsvinay0717No ratings yet
- Drug Development FunnelDocument2 pagesDrug Development FunnelSALEHA HASSANNo ratings yet
- Clinical StudiesDocument14 pagesClinical StudiesDWALE AUBADENo ratings yet
- New Drug Application - WikipediaDocument26 pagesNew Drug Application - Wikipediakabirsahu0019No ratings yet
- Organizational Plan of Pharmaceutical CompanyDocument5 pagesOrganizational Plan of Pharmaceutical Companynoorehira2929No ratings yet
- clinical trialsDocument5 pagesclinical trialskamwebazemarkkNo ratings yet
- 26 Spruit DragonairDocument2 pages26 Spruit DragonairW.J. ZondagNo ratings yet
- IET Biometrics - 2022 - Sun - Breast Mass Classification Based On Supervised Contrastive Learning and Multi ViewDocument13 pagesIET Biometrics - 2022 - Sun - Breast Mass Classification Based On Supervised Contrastive Learning and Multi ViewMaha MasNo ratings yet
- Phy CIA 2mDocument25 pagesPhy CIA 2mA/E5LOGESH AKRNo ratings yet
- Topic 6 Circular Motion Test P1Document11 pagesTopic 6 Circular Motion Test P1Sabrina_LGXNo ratings yet
- Addition of Green Tea Extract in Fructose Egg Yolk Milk Diluter On The Quality and Antioxidant ContenDocument9 pagesAddition of Green Tea Extract in Fructose Egg Yolk Milk Diluter On The Quality and Antioxidant Conten3129 NashihahNo ratings yet
- SMA 2370 - CALCULUS IV - Main Campus-PRINTREADYDocument3 pagesSMA 2370 - CALCULUS IV - Main Campus-PRINTREADYEVANS KIPNGETICHNo ratings yet
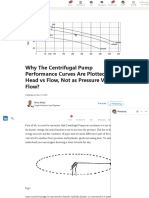
- Why The Centrifugal Pump Performance Curves Are Plotted As Head Vs Flow, Not As Pressure Versus Flow - LinkedInDocument8 pagesWhy The Centrifugal Pump Performance Curves Are Plotted As Head Vs Flow, Not As Pressure Versus Flow - LinkedInJitendraHatwarNo ratings yet
- C-Question-Bank-ebook VIPSDocument46 pagesC-Question-Bank-ebook VIPSlalit sainiNo ratings yet
- 10 Most Powerful Greek GodsDocument4 pages10 Most Powerful Greek GodsIris Milagro MembreñoNo ratings yet
- 10 Herbal Medicines Approved by The DohDocument13 pages10 Herbal Medicines Approved by The DohLark SantiagoNo ratings yet
- ODME FailureDocument3 pagesODME FailureNatalie JonesNo ratings yet
- A Crisis Intervention Model For Child Protective S PDFDocument22 pagesA Crisis Intervention Model For Child Protective S PDFKali HartvigsenNo ratings yet
- Westermo Ds Ddw-x42-Series 2204 en RevfDocument5 pagesWestermo Ds Ddw-x42-Series 2204 en RevfAnanthan GunasaharanNo ratings yet
- Kinoton FP30D Operating ManualDocument87 pagesKinoton FP30D Operating ManualTomekLecocqNo ratings yet
- Led Music Controller InsructionsDocument2 pagesLed Music Controller InsructionsArnaldoloNo ratings yet
- Chemistry Project PDFDocument30 pagesChemistry Project PDFyash hiraniNo ratings yet
- 1Document11 pages1putriNo ratings yet
- Turbine Cat SolarDocument2 pagesTurbine Cat SolarAbid Lakhani100% (1)
- High Voltage Testing Measurement Equipment CatalogDocument46 pagesHigh Voltage Testing Measurement Equipment CatalogHari HidayatNo ratings yet
- Book's SolutionsDocument20 pagesBook's SolutionsKodjo ALIPUINo ratings yet
- Module 5 Ge Elec 3Document10 pagesModule 5 Ge Elec 3Crishell Hunahunan AngelesNo ratings yet
- JSW New Final ProjetDocument48 pagesJSW New Final ProjetRajender SinghNo ratings yet
- SP-1102A Specification For Design of 33kV Overhead Power Lines On Wooden PolesDocument118 pagesSP-1102A Specification For Design of 33kV Overhead Power Lines On Wooden Polesarjunprasannan7No ratings yet
- Abe 424 Farm Structures and Environmental ControlDocument42 pagesAbe 424 Farm Structures and Environmental ControlAmabi SilasNo ratings yet
- The Baldur's Gate Series 2 - Shadows of AmnDocument99 pagesThe Baldur's Gate Series 2 - Shadows of AmnJustin Moore100% (1)
- Wipro Ge Healthcare Private Limited PDFDocument13 pagesWipro Ge Healthcare Private Limited PDFMandal TusharNo ratings yet
- Ebook Ebook PDF Principles of Human Physiology 6th Edition PDFDocument41 pagesEbook Ebook PDF Principles of Human Physiology 6th Edition PDFdonita.nichols650100% (43)