Download as pdf or txt
You might also like
- Sbi Mains Admit CardDocument2 pagesSbi Mains Admit CardnavneetNo ratings yet
- Statistics for Bioinformatics: Methods for Multiple Sequence AlignmentFrom EverandStatistics for Bioinformatics: Methods for Multiple Sequence AlignmentNo ratings yet
- In Silico Attributes of Cold Resistance Protein 1 (Crp1) F Rom Brassica OleraceaeDocument8 pagesIn Silico Attributes of Cold Resistance Protein 1 (Crp1) F Rom Brassica OleraceaeresearchinventyNo ratings yet
- 2015 Article 14 Twilight ZoneDocument11 pages2015 Article 14 Twilight Zoneadiav PakNo ratings yet
- 2013 J Theor Biol 328 77-88Document12 pages2013 J Theor Biol 328 77-88mbrylinskiNo ratings yet
- I-TASSER: A Unified Platform For Automated Protein Structure and Function PredictionDocument14 pagesI-TASSER: A Unified Platform For Automated Protein Structure and Function PredictionDavidNo ratings yet
- Unit 1: Structural GenomicsDocument4 pagesUnit 1: Structural GenomicsLavanya ReddyNo ratings yet
- Mol - Modelling of BCL-2 FinalDocument37 pagesMol - Modelling of BCL-2 FinalVannakumar SubramanianNo ratings yet
- 2010 Proteins 78 118-134Document17 pages2010 Proteins 78 118-134mbrylinskiNo ratings yet
- MP-T Improving Membrane Protein Alignment For StructureDocument8 pagesMP-T Improving Membrane Protein Alignment For StructureCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- The Role of Protein Structure in Genomics: MinireviewDocument5 pagesThe Role of Protein Structure in Genomics: MinireviewAakriti SharmaNo ratings yet
- A Topology-Based Network Tree For The Prediction of Protein-Protein Binding Affinity Changes Following MutationDocument8 pagesA Topology-Based Network Tree For The Prediction of Protein-Protein Binding Affinity Changes Following Mutation1813975862yvyvNo ratings yet
- Gao HPC-ML FinalDocument12 pagesGao HPC-ML FinalAda SedovaNo ratings yet
- Protein Unfolding Behavior Studied by Elastic Network ModelDocument11 pagesProtein Unfolding Behavior Studied by Elastic Network ModeljmxnetdjNo ratings yet
- HPCINDocument13 pagesHPCINdev_sNo ratings yet
- COACH YZhangDocument8 pagesCOACH YZhangChristos FeidakisNo ratings yet
- Integrative Modeling of Membrane-Associated Protein AssembliesDocument12 pagesIntegrative Modeling of Membrane-Associated Protein AssembliesConstanza IsabellaNo ratings yet
- Homo LogyDocument8 pagesHomo LogyIgnacioEguinoaNo ratings yet
- 10.IJAEST Vol No 7 Issue No 2 Human Protein Function Prediction An Overview 239 244Document6 pages10.IJAEST Vol No 7 Issue No 2 Human Protein Function Prediction An Overview 239 244helpdesk9532No ratings yet
- Graph Based SignatureDocument8 pagesGraph Based SignatureNyasha ChitavatiNo ratings yet
- Cyclic Peptide Structure Prediction and Design Using AlphaFoldDocument25 pagesCyclic Peptide Structure Prediction and Design Using AlphaFoldxinyuwu0528No ratings yet
- Bioinformatics 33-22-3549Document9 pagesBioinformatics 33-22-3549donkingdedon30No ratings yet
- 1 s2.0 S2452310017300926 MainDocument9 pages1 s2.0 S2452310017300926 MainBen DresimNo ratings yet
- Stam 2004Document12 pagesStam 2004CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Classification of Protein-Protein Association Rates Based On Biophysical InformaticsDocument20 pagesClassification of Protein-Protein Association Rates Based On Biophysical InformaticsMe KhNo ratings yet
- Prediction of Protein Secondary Structure With A Reliability Score Estimated by Local Sequence ClusteringDocument7 pagesPrediction of Protein Secondary Structure With A Reliability Score Estimated by Local Sequence ClusteringShampa SenNo ratings yet
- Bioinformatics AnswersDocument13 pagesBioinformatics AnswersPratibha Patil100% (1)
- 2009 Brief Bioinform 10 378-391Document14 pages2009 Brief Bioinform 10 378-391mbrylinskiNo ratings yet
- Generative Modeling For Protein StructuresDocument12 pagesGenerative Modeling For Protein Structurescarlos ArozamenaNo ratings yet
- Proteo MicsDocument25 pagesProteo MicsSaptarshi GhoshNo ratings yet
- Hard WiredDocument9 pagesHard WiredHNNo ratings yet
- TMP D73Document15 pagesTMP D73FrontiersNo ratings yet
- Image-Based Spatiotemporal Causality Inference For Protein Signaling NetworksDocument8 pagesImage-Based Spatiotemporal Causality Inference For Protein Signaling Networksmtallis3142275No ratings yet
- Opportunities and Challenges For Target Identification and Lead Structural Biology and Bioinformatics in Drug DesignDocument12 pagesOpportunities and Challenges For Target Identification and Lead Structural Biology and Bioinformatics in Drug DesignpavanapexNo ratings yet
- Basics of ProteomicsDocument7 pagesBasics of ProteomicskonndaaiahNo ratings yet
- Statistical Measures To Quantify Similarity Between Molecular Dynamics Simulation TrajectoriesDocument17 pagesStatistical Measures To Quantify Similarity Between Molecular Dynamics Simulation TrajectoriesDiego GranadosNo ratings yet
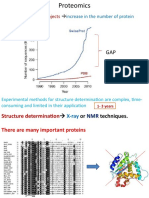
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- Structural Biology On Mac OS X: Technology, Tools, and WorkflowsDocument29 pagesStructural Biology On Mac OS X: Technology, Tools, and WorkflowsLê HànNo ratings yet
- Dynamics RelateDocument11 pagesDynamics RelatevijayNo ratings yet
- Structural BioinfoDocument76 pagesStructural BioinfoMudit MisraNo ratings yet
- Protein Structure Prediction ThesisDocument8 pagesProtein Structure Prediction ThesisWriteMyThesisPaperManchester100% (2)
- On The Accuracy of Homology Modeling and Sequence AlignmentDocument10 pagesOn The Accuracy of Homology Modeling and Sequence AlignmentCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Prediction of Protein-Protein Interactions With LSTM Deep Learning ModelDocument5 pagesPrediction of Protein-Protein Interactions With LSTM Deep Learning ModelAfaque AlamNo ratings yet
- Proteins: Remote Homology Detection of Integral Membrane Proteins Using Conserved Sequence FeaturesDocument13 pagesProteins: Remote Homology Detection of Integral Membrane Proteins Using Conserved Sequence FeaturesLavanya Priya SathyanNo ratings yet
- DeepAffinity: Interpretable Deep Learning of Compound-Protein Affinity Through Unified Recurrent and Convolutional Neural NetworksDocument10 pagesDeepAffinity: Interpretable Deep Learning of Compound-Protein Affinity Through Unified Recurrent and Convolutional Neural Networkswangxiangwen0201No ratings yet
- 2023 02 05 527174v1 FullDocument24 pages2023 02 05 527174v1 FullpucchucNo ratings yet
- 2011 J Struct Biol 173 558-569Document12 pages2011 J Struct Biol 173 558-569mbrylinskiNo ratings yet
- Sali Structure 2003Document5 pagesSali Structure 2003hahahaNo ratings yet
- 1 s2.0 S2950347724000239 MainDocument30 pages1 s2.0 S2950347724000239 MainAnonno Singha RayNo ratings yet
- Cbar: A Computer Program To Distinguish Plasmid-Derived From Chromosome-Derived Sequence Fragments in Metagenomics DataDocument2 pagesCbar: A Computer Program To Distinguish Plasmid-Derived From Chromosome-Derived Sequence Fragments in Metagenomics Datadumboo21No ratings yet
- Journal of Bioinformatics and Computational Biology Vol. 10, No. 4 (2012) 1203002 (3 Pages) C Imperial College Press DoiDocument3 pagesJournal of Bioinformatics and Computational Biology Vol. 10, No. 4 (2012) 1203002 (3 Pages) C Imperial College Press DoijimakosjpNo ratings yet
- 2010 Bioinformatics 26 687-688Document2 pages2010 Bioinformatics 26 687-688mbrylinskiNo ratings yet
- 1 s2.0 S1367593121000600 MainDocument7 pages1 s2.0 S1367593121000600 MainSumaiya Sadia BristyNo ratings yet
- 2019, Multifaceted Protein-Protein Interaction Prediction Based On Siamese Residual RCNNDocument10 pages2019, Multifaceted Protein-Protein Interaction Prediction Based On Siamese Residual RCNNAfaque AlamNo ratings yet
- Research Papers On Protein FoldingDocument5 pagesResearch Papers On Protein Foldingshvximwhf100% (1)
- A Boolean-Based Systems Biology Approach To Predict Novel Genes Associated With Cancer: Application To Colorectal CancerDocument15 pagesA Boolean-Based Systems Biology Approach To Predict Novel Genes Associated With Cancer: Application To Colorectal CancerAries YuanggaNo ratings yet
- Algorithms and Methods in Structural Bioinformatics (Computational Biology) (Nurit Haspel (Editor) Etc.)Document120 pagesAlgorithms and Methods in Structural Bioinformatics (Computational Biology) (Nurit Haspel (Editor) Etc.)Sathish KumarNo ratings yet
- Multifaceted Protein-Protein Interaction Prediction Based On Siamese Residual RCNNDocument10 pagesMultifaceted Protein-Protein Interaction Prediction Based On Siamese Residual RCNNAfaque AlamNo ratings yet
- Not Every Estimate Counts - Evaluation of Cell Composition Estimation Approaches in Brain Bulk Tissue DataDocument14 pagesNot Every Estimate Counts - Evaluation of Cell Composition Estimation Approaches in Brain Bulk Tissue DataSergio VillicañaNo ratings yet
- Muito Detalhada Essa Revisão TranscriptomicaDocument14 pagesMuito Detalhada Essa Revisão TranscriptomicaLeticia PontesNo ratings yet
- Boombot ProDocument13 pagesBoombot Proriadh.bachwellNo ratings yet
- K-Bglu 1107 DataDocument16 pagesK-Bglu 1107 Datapi_yoanaNo ratings yet
- MS-CIT Question Bank 1: Answer Start ButtonDocument29 pagesMS-CIT Question Bank 1: Answer Start ButtonChamika Madushan ManawaduNo ratings yet
- Servicenow Application Developer Exam New-Practice Test Set 2Document30 pagesServicenow Application Developer Exam New-Practice Test Set 2Apoorv DiwanNo ratings yet
- Irrigation Engineering: Course Instructor: Engr. Arif Asghar GopangDocument22 pagesIrrigation Engineering: Course Instructor: Engr. Arif Asghar GopangArif AsgharNo ratings yet
- Get Started On Tumblr in 5 Easy StepsDocument7 pagesGet Started On Tumblr in 5 Easy Stepspolarbear3No ratings yet
- Software Engineer ResumeDocument1 pageSoftware Engineer ResumepareekrachitNo ratings yet
- Dwnload Full Intermediate Microeconomics and Its Application 11th Edition Nicholson Solutions Manual PDFDocument36 pagesDwnload Full Intermediate Microeconomics and Its Application 11th Edition Nicholson Solutions Manual PDFmateopnpowell100% (13)
- EMC Standard PDFDocument58 pagesEMC Standard PDFSongkran Pisanupoj0% (1)
- Manual Indonesia Ford MondeoDocument197 pagesManual Indonesia Ford MondeoCha-cha ThaLiefNo ratings yet
- Effects of Nanosilica Additions On Cement Pastes: A Porro J S Dolado I Campillo E Erkizia Y de Miguel Y Saez de IbarraDocument10 pagesEffects of Nanosilica Additions On Cement Pastes: A Porro J S Dolado I Campillo E Erkizia Y de Miguel Y Saez de IbarraMariwan Mirhaj MohamedSalihNo ratings yet
- Construction Safety - Part 2 (Site Premises)Document20 pagesConstruction Safety - Part 2 (Site Premises)Henry Turalde0% (1)
- Chapter Two Training and DevelopmentDocument13 pagesChapter Two Training and DevelopmentMonira Mohamed YousufNo ratings yet
- Bill Presentment ArchitectureDocument33 pagesBill Presentment ArchitecturevenkatNo ratings yet
- Project Gant Chart & Cost S-Curve-OriginalDocument2 pagesProject Gant Chart & Cost S-Curve-OriginalMuhammad Anamul HoqueNo ratings yet
- Differences Between Hypertext, Hypermedia and MultimediaDocument7 pagesDifferences Between Hypertext, Hypermedia and MultimediaEdison Alarcón90% (20)
- A Theoretical Framework of Flood InducedDocument5 pagesA Theoretical Framework of Flood InducedPete PuertoNo ratings yet
- Business Transactions Their AnalysisDocument5 pagesBusiness Transactions Their AnalysisJeah PanganibanNo ratings yet
- The Latest Thing in Black and White - Smart Colour CapabilityDocument12 pagesThe Latest Thing in Black and White - Smart Colour CapabilityAhmad YousefNo ratings yet
- Payslip - 12.09.2021Document1 pagePayslip - 12.09.2021Madison MooreNo ratings yet
- Veova10 - MSDSDocument8 pagesVeova10 - MSDSMohamed HalemNo ratings yet
- CH 8Document14 pagesCH 8Ajay Kumar GangulyNo ratings yet
- Compiled ProjectDocument74 pagesCompiled ProjectMISHEAL CHIEMELANo ratings yet
- Cognos ResumeDocument4 pagesCognos ResumeAnonymous xMYE0TiNBcNo ratings yet
- Analysis of Thermal Behavior of High Frequency Transformers Using Finite Element MethodDocument6 pagesAnalysis of Thermal Behavior of High Frequency Transformers Using Finite Element Methodkar mugilanNo ratings yet
- UDP Message FormatDocument5 pagesUDP Message FormatGauravNo ratings yet
- Psychology of RehabilitationDocument154 pagesPsychology of RehabilitationDhruv JainNo ratings yet
- Examination Procedures For Vacuum TestDocument9 pagesExamination Procedures For Vacuum TestOsilonya HenryNo ratings yet
- Human Retention Using Data ScienceDocument16 pagesHuman Retention Using Data ScienceDivya PatilNo ratings yet