Download as pdf or txt
You might also like
- Vat Receipt: MR Kyle BoydDocument1 pageVat Receipt: MR Kyle BoydNovak Brodsky83% (6)
- Biopharmaceutical Classification System and Formulation DevelopmentDocument18 pagesBiopharmaceutical Classification System and Formulation DevelopmentAshish Mittal100% (1)
- BCS WHO Classification QAS04 109rev1 Waive Invivo BioequivDocument45 pagesBCS WHO Classification QAS04 109rev1 Waive Invivo BioequivjjmuruzaNo ratings yet
- Biopharmaceutics Classification System: Systematic Reviews in Pharmacy January 2010Document9 pagesBiopharmaceutics Classification System: Systematic Reviews in Pharmacy January 2010afdsafNo ratings yet
- BCS ClassDocument8 pagesBCS Classrina wijayantiNo ratings yet
- Biopharmaceutics Classification System: Review ArticleDocument8 pagesBiopharmaceutics Classification System: Review ArticleAileen AngNo ratings yet
- Annex 8: Proposal To Waive in Vivo Bioequivalence Requirements For WHO Model List of EssentialDocument49 pagesAnnex 8: Proposal To Waive in Vivo Bioequivalence Requirements For WHO Model List of Essentialrnd_holiNo ratings yet
- S19640en PDFDocument47 pagesS19640en PDFDrSyeda RimaNo ratings yet
- Bcs JurnalDocument7 pagesBcs JurnalRny_SKNo ratings yet
- Biopharmaceutical Classification System An Account-1Document11 pagesBiopharmaceutical Classification System An Account-1Gia BarusNo ratings yet
- Biopharmaceutics Classification SystemDocument3 pagesBiopharmaceutics Classification SystemAnti MariantiNo ratings yet
- Biowaivers Criteria RequirementsDocument11 pagesBiowaivers Criteria RequirementsSaravanan AnnamalaiNo ratings yet
- BCS REg ApproachDocument7 pagesBCS REg Approachvg_vvgNo ratings yet
- Biopharmaceutics Classification System-The Scientific Basis PDFDocument5 pagesBiopharmaceutics Classification System-The Scientific Basis PDFtrianawidiacandra100% (1)
- Itle: Dissolution Testing 1 2Document16 pagesItle: Dissolution Testing 1 2hassaan haiderNo ratings yet
- Overview of Factors Affecting Oral Drug AbsorptionDocument11 pagesOverview of Factors Affecting Oral Drug Absorptionmahveen balochNo ratings yet
- Update On Gastrointestinal Biorelevant Media and Physiologically Relevant Dissolution ConditionsDocument14 pagesUpdate On Gastrointestinal Biorelevant Media and Physiologically Relevant Dissolution ConditionsVirajNo ratings yet
- Cristofoletti2013 PDFDocument9 pagesCristofoletti2013 PDFShinta LestariNo ratings yet
- Biopharmaceutical Classification System: A Strategic Tool For Oral Drug Delivery TechnologyDocument6 pagesBiopharmaceutical Classification System: A Strategic Tool For Oral Drug Delivery TechnologyHijrawati Ayu WardaniNo ratings yet
- Biopharmaceutical Classification System: A Strategic Tool For Oral Drug Delivery TechnologyDocument6 pagesBiopharmaceutical Classification System: A Strategic Tool For Oral Drug Delivery TechnologyAgus PriyonoNo ratings yet
- BCSDocument10 pagesBCSruchit_darjiNo ratings yet
- El Rol Del Sistema de Clasificación BiofarmacéuticaDocument3 pagesEl Rol Del Sistema de Clasificación BiofarmacéuticaMaria Cecilia Morcillo MuñozNo ratings yet
- Chicken 7Document10 pagesChicken 7Mary VegaNo ratings yet
- 10 1016@j Ijpharm 2019 05 041Document47 pages10 1016@j Ijpharm 2019 05 041Shinta LestariNo ratings yet
- BCS - Is The Full Potential Reached 2014Document8 pagesBCS - Is The Full Potential Reached 2014ivaNo ratings yet
- Biowaiver: An Alternative To in Vivo Pharmacokinetic Bioequivalence StudiesDocument8 pagesBiowaiver: An Alternative To in Vivo Pharmacokinetic Bioequivalence StudiesVinz AlvarezNo ratings yet
- Classification of Orally Administered Drugs On The World Health Organization Model List of Essential Medicines According To The Bio Pharmaceutics Classification SystemDocument14 pagesClassification of Orally Administered Drugs On The World Health Organization Model List of Essential Medicines According To The Bio Pharmaceutics Classification SystemEr'el AR100% (1)
- BCS Class PDFDocument14 pagesBCS Class PDFdhimas11100% (1)
- Assignment I SolubilityDocument24 pagesAssignment I Solubilityabthapa100% (1)
- 01 Shah FDA Pqri Conf Bcs 15Document17 pages01 Shah FDA Pqri Conf Bcs 15Karnhia RamadhanNo ratings yet
- Penting BcsDocument26 pagesPenting BcsDEVIANo ratings yet
- The Use of Biopharmaceutic Classification of Drugs in Drug Discovery and Development: Current Status and Future ExtensionDocument13 pagesThe Use of Biopharmaceutic Classification of Drugs in Drug Discovery and Development: Current Status and Future ExtensionMateo BenítezNo ratings yet
- BCS Classification ArticleDocument17 pagesBCS Classification ArticleHARI HARA RAO GUJJARNo ratings yet
- Tsume 2014Document46 pagesTsume 2014Juan PerezNo ratings yet
- trs1019 Annex4 Classification System Based Classification of Active Pharmaceutical Ingredients For BiowaiverDocument16 pagestrs1019 Annex4 Classification System Based Classification of Active Pharmaceutical Ingredients For Biowaiverايناس ماجدNo ratings yet
- Pharmaceutics 15 00753Document16 pagesPharmaceutics 15 00753Fabio GreenNo ratings yet
- Internal Assigment LiquidDocument6 pagesInternal Assigment LiquidRoa'a Abu arqoobNo ratings yet
- Bioavailability and BioequivalenceDocument28 pagesBioavailability and Bioequivalencekulbhushan singhNo ratings yet
- s12248 022 00687 0Document17 pagess12248 022 00687 0Lina WinartiNo ratings yet
- The Role of BCS (Biopharmaceutics Classification System) and BDDCS (Biopharmaceutics Drug Disposition Classification System) in Drug Development Need To Be PrintedDocument9 pagesThe Role of BCS (Biopharmaceutics Classification System) and BDDCS (Biopharmaceutics Drug Disposition Classification System) in Drug Development Need To Be PrintedKimberly MccoyNo ratings yet
- Biowaiver Monographs For Immediate Release Solid Oral Dosage Forms: Acetaminophen (Paracetamol)Document13 pagesBiowaiver Monographs For Immediate Release Solid Oral Dosage Forms: Acetaminophen (Paracetamol)Candy CarlaNo ratings yet
- Selection of DissolutionDocument5 pagesSelection of DissolutionGirishNo ratings yet
- Pharmaceutics 14 02565Document12 pagesPharmaceutics 14 02565Sebas IsraelNo ratings yet
- Intrinsic DissolutionDocument8 pagesIntrinsic DissolutionMathaios VadilisNo ratings yet
- Oral Drug Absorption and The Biopharmaceutics Classification SystemDocument8 pagesOral Drug Absorption and The Biopharmaceutics Classification SystemyanuarNo ratings yet
- Art 00001Document8 pagesArt 00001Geo GeoNo ratings yet
- Biorelevant Dissolution: Methodology and Application in Drug DevelopmentDocument7 pagesBiorelevant Dissolution: Methodology and Application in Drug DevelopmentPedro MaiaNo ratings yet
- BiopharmaceuticalClassificationSystem PDFDocument7 pagesBiopharmaceuticalClassificationSystem PDFShahd Diefalla Ahmed DiefallaNo ratings yet
- Prakash 2Document2 pagesPrakash 2Patel PrakashkumarNo ratings yet
- Bio Pharmaceutical Classification SystemDocument7 pagesBio Pharmaceutical Classification SystemRafi Arif 7189No ratings yet
- Chapter 9: Solid Oral Modified-Release Dosage Forms and Drug Delivery SystemsDocument26 pagesChapter 9: Solid Oral Modified-Release Dosage Forms and Drug Delivery SystemsAlecza Mae Savella100% (1)
- Guidelines For Bioavailability and Bioequivalence Studies: A ReviewDocument6 pagesGuidelines For Bioavailability and Bioequivalence Studies: A ReviewNagabhushanam MaddiNo ratings yet
- Bcs Classification of DrugsDocument12 pagesBcs Classification of Drugsjigarpatel5No ratings yet
- Review Article VikashdashDocument4 pagesReview Article VikashdashamjohnnyNo ratings yet
- Predict WordDocument13 pagesPredict Wordtata permataNo ratings yet
- In Silico Bcs For Oral Drug DevelopmentDocument13 pagesIn Silico Bcs For Oral Drug DevelopmentShin KaojuNo ratings yet
- LA - BCS M9 Guidance Training - FINALspDocument31 pagesLA - BCS M9 Guidance Training - FINALspErnesto AnayaNo ratings yet
- BCS Class of DrugsDocument45 pagesBCS Class of DrugsLionO50% (2)
- A Comprehensive Book on Experimental PharmaceuticsFrom EverandA Comprehensive Book on Experimental PharmaceuticsRating: 5 out of 5 stars5/5 (1)
- Physicochemical and Biomimetic Properties in Drug Discovery: Chromatographic Techniques for Lead OptimizationFrom EverandPhysicochemical and Biomimetic Properties in Drug Discovery: Chromatographic Techniques for Lead OptimizationNo ratings yet
- By Robert B Salter Textbook of Disorders and Injuries of The Musculoskeletal System Third 3rd Edition by Author B004hgvxko PDFDocument5 pagesBy Robert B Salter Textbook of Disorders and Injuries of The Musculoskeletal System Third 3rd Edition by Author B004hgvxko PDFDita ayu0% (2)
- Peripheral Blood Smear Pathologist ToolDocument3 pagesPeripheral Blood Smear Pathologist ToolSimon HafeniNo ratings yet
- Badminton Is A Court or Lawn Game Played With Lightweight Racketsand AshuttlecockDocument14 pagesBadminton Is A Court or Lawn Game Played With Lightweight Racketsand Ashuttlecocktaehyung trash100% (1)
- Paan TobaccoDocument8 pagesPaan TobaccoZaibunnisa WasiqNo ratings yet
- Optimisation of Supply Chain of Smart ColoursDocument29 pagesOptimisation of Supply Chain of Smart ColoursPooja ShahNo ratings yet
- FFXV Checklist - HuntsDocument1 pageFFXV Checklist - HuntsarashibirruNo ratings yet
- PO Lifecycle in SAPDocument76 pagesPO Lifecycle in SAPadwankarparagNo ratings yet
- Experiment No. (6) Study of Dispersion Compensation Schemes: ObjectDocument9 pagesExperiment No. (6) Study of Dispersion Compensation Schemes: ObjectFaez FawwazNo ratings yet
- Intermolecular Forces of Liquids and Solids Solids and Their Properties PDFDocument13 pagesIntermolecular Forces of Liquids and Solids Solids and Their Properties PDFpieNo ratings yet
- Tugas GinjalDocument22 pagesTugas GinjalAnastasia MargaretNo ratings yet
- Fce StrategiesDocument19 pagesFce StrategiesTere AguirreNo ratings yet
- Courtyard: Unlucky Tabibito ( )Document1 pageCourtyard: Unlucky Tabibito ( )Wraith VoidNo ratings yet
- Ensemble LearningDocument7 pagesEnsemble LearningJavier Garcia RajoyNo ratings yet
- Step 5 and 6Document6 pagesStep 5 and 6Diana Rose MitoNo ratings yet
- Holding Companies: Problems and Solutions - AccountingDocument17 pagesHolding Companies: Problems and Solutions - AccountingVaibhav MaheshwariNo ratings yet
- Personal Branding Workbook - Ext - 2nd EdDocument23 pagesPersonal Branding Workbook - Ext - 2nd EdMiguelito JeromeNo ratings yet
- Vxflex Data SheetDocument5 pagesVxflex Data SheetJesus SantiagoNo ratings yet
- SF15 XCM DS RL 29 80,100Document1 pageSF15 XCM DS RL 29 80,100Vagner PaludoNo ratings yet
- NCP IrritabilityDocument3 pagesNCP IrritabilityBruce Kelly MamarilNo ratings yet
- B P S U: Ataan Eninsula Tate NiversityDocument10 pagesB P S U: Ataan Eninsula Tate NiversityCharles Vincent DavidNo ratings yet
- Moves That Make You Feel Great: Calf Stretch Chair SquatDocument2 pagesMoves That Make You Feel Great: Calf Stretch Chair Squatnotsosafe89No ratings yet
- GR9 - Q4 - Lesson 1 - Motion in One DimensionDocument5 pagesGR9 - Q4 - Lesson 1 - Motion in One DimensionJerome HizonNo ratings yet
- BBRC4103 Kaedah Penyelidikan September Semester 2020Document12 pagesBBRC4103 Kaedah Penyelidikan September Semester 2020SARIPAH NOOR FARAH AIN BINTI P.AFDZAL SHAH STUDENTNo ratings yet
- Parker Valvula Direcional D3W PDFDocument20 pagesParker Valvula Direcional D3W PDFChagas OliveiraNo ratings yet
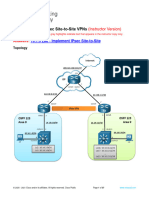
- 16.1.3 Lab - Implement IPsec Site-to-Site - ITExamAnswersDocument37 pages16.1.3 Lab - Implement IPsec Site-to-Site - ITExamAnswershayltonmonteiroNo ratings yet
- Basic Customer Service Principles-ImportantDocument39 pagesBasic Customer Service Principles-Importantzoltan2014100% (6)
- PrelimsDocument1 pagePrelimsCristina LaniohanNo ratings yet
- Sri Mahila Griha Udyog Lijjat Papad: Strategic Management CourseDocument23 pagesSri Mahila Griha Udyog Lijjat Papad: Strategic Management CoursePrachit ChaturvediNo ratings yet
- 1 The Building Blocks of JapanDocument26 pages1 The Building Blocks of JapanschelleslarsNo ratings yet