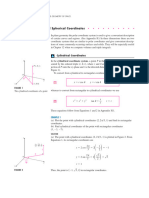
Download as pdf or txt
You might also like
- 2nd Puc Maths Sulalitha Exam Practice Guide Eng Version 2020-21 by ChikkaballapuraDocument101 pages2nd Puc Maths Sulalitha Exam Practice Guide Eng Version 2020-21 by Chikkaballapurabot insaan82% (90)
- Crystal StructureDocument6 pagesCrystal StructureAlfian GilangNo ratings yet
- Omar ShabanaDocument12 pagesOmar ShabanaCleves AxiomaNo ratings yet
- Circular Functions: SectionDocument10 pagesCircular Functions: SectionFlonie DensingNo ratings yet
- Reciprocal Space PDFDocument63 pagesReciprocal Space PDFOhidul IslamNo ratings yet
- Knot Spec RoundDocument28 pagesKnot Spec RoundSusila WindartaNo ratings yet
- Target Paper XIDocument5 pagesTarget Paper XIAlizaaNo ratings yet
- Miller Indices: Planes DirectionsDocument47 pagesMiller Indices: Planes DirectionsSilvers Rayleigh100% (1)
- Go KaranDocument12 pagesGo KaranGokaranNo ratings yet
- IJMEIT Kudin Sandwich1 PDFDocument6 pagesIJMEIT Kudin Sandwich1 PDFavk256No ratings yet
- A0603010108 PDFDocument8 pagesA0603010108 PDFtheijesNo ratings yet
- Zapata Combinada Joseph E. Bowles Analytical and Computer Methods in Foundation EngineeringDocument14 pagesZapata Combinada Joseph E. Bowles Analytical and Computer Methods in Foundation Engineeringlogos7100% (1)
- Chapter 2 Geometry of CrystalsDocument56 pagesChapter 2 Geometry of CrystalssfdafNo ratings yet
- Formation of Y-Bus MatrixDocument3 pagesFormation of Y-Bus Matrixsanjay AdhikariNo ratings yet
- I. Vectors and Geometry in Two and Three DimensionsDocument34 pagesI. Vectors and Geometry in Two and Three DimensionsAkpa KenechukwuNo ratings yet
- STINY Ice-RayDocument10 pagesSTINY Ice-RayjerryNo ratings yet
- Use E-Papers, Save Trees: Above Line Hide When Print OutDocument101 pagesUse E-Papers, Save Trees: Above Line Hide When Print OutPRUTHVI100% (2)
- Problems: 2-1 Show The Transmission and Deviation Angles For The Two Mechanisms, Taking The Input To BeDocument34 pagesProblems: 2-1 Show The Transmission and Deviation Angles For The Two Mechanisms, Taking The Input To BeNegar SatvatNo ratings yet
- 2014 N5 Maths P2Document14 pages2014 N5 Maths P2kamraanryanNo ratings yet
- Polinomios de RookDocument7 pagesPolinomios de RookDanielValadãoBastosNo ratings yet
- S PDFDocument15 pagesS PDFChitsimran Singh AroraNo ratings yet
- Analysis of Transmission Electron Microscopy Contrast at Extrinsic Grain Boundary Doslocations Bu R.A. VarinDocument12 pagesAnalysis of Transmission Electron Microscopy Contrast at Extrinsic Grain Boundary Doslocations Bu R.A. VarinSamuel ManaluNo ratings yet
- Condensed Matter Physics Unit 1Document112 pagesCondensed Matter Physics Unit 1anujjuetNo ratings yet
- 2018 普物II 1stDocument13 pages2018 普物II 1styulin linNo ratings yet
- Adjustment of Cable - Stayed Bridges:, IncludingDocument11 pagesAdjustment of Cable - Stayed Bridges:, IncludingVijay KumarNo ratings yet
- Lattices DiffractionDocument20 pagesLattices DiffractionShafiqul Islam MahfuzNo ratings yet
- Assignment No.4Document3 pagesAssignment No.4Abhishek NarwariyaNo ratings yet
- Assignment No.4Document3 pagesAssignment No.422mc10sh584No ratings yet
- Quantum Mechanics - Last 5 Years Slet / Net Question Bank: Institute For TRB, Slet and Net in PhysicsDocument62 pagesQuantum Mechanics - Last 5 Years Slet / Net Question Bank: Institute For TRB, Slet and Net in Physics22pgpe16No ratings yet
- LatticeDocument59 pagesLatticeashishkr23438No ratings yet
- Khoa học Bề mặt Chất rắnDocument25 pagesKhoa học Bề mặt Chất rắnVĩnh Khoa NgôNo ratings yet
- LatticeDocument56 pagesLatticeashok pradhanNo ratings yet
- 2-Geometry of Crystals II Reciprocal Lattice and SymmetryDocument61 pages2-Geometry of Crystals II Reciprocal Lattice and Symmetry李震原No ratings yet
- Computer Graphics Forum - 2003 - Stam - Quad Triangle SubdivisionDocument7 pagesComputer Graphics Forum - 2003 - Stam - Quad Triangle SubdivisionMpampisTsatsosNo ratings yet
- EX0703Document3 pagesEX0703igualdi53No ratings yet
- Chapter 2 - Applications of Group Theory 2.1 How Group Theory Applies To A Variety of Chemical ProblemsDocument16 pagesChapter 2 - Applications of Group Theory 2.1 How Group Theory Applies To A Variety of Chemical ProblemsRameshkumarNo ratings yet
- Vectors 1Document5 pagesVectors 1aanya.panditNo ratings yet
- Matrix MathDocument20 pagesMatrix MathHadi Hassan100% (1)
- Centroids and Centers of GravityDocument4 pagesCentroids and Centers of GravitycarminavegamariaNo ratings yet
- 08AL PHYessayDocument20 pages08AL PHYessayAhmad HasanNo ratings yet
- C1 - Basics of VectorsDocument13 pagesC1 - Basics of VectorsCheche TiberiusNo ratings yet
- 4.13C Huckel MO TheoryDocument6 pages4.13C Huckel MO TheoryHaroon RashidNo ratings yet
- Home Work Solutions 9Document5 pagesHome Work Solutions 9satryaNo ratings yet
- Demoivres TheoremDocument10 pagesDemoivres TheoremExcel Edson ChibaNo ratings yet
- Reciprocal Lattice & Brillouin ZoneDocument20 pagesReciprocal Lattice & Brillouin ZoneAkshay KumarNo ratings yet
- Cylindrical Coordinates: D.1 Local Basis VectorsDocument4 pagesCylindrical Coordinates: D.1 Local Basis VectorsAraz CabbarlıNo ratings yet
- Unit 3 Keplers LawDocument3 pagesUnit 3 Keplers LawSagar ChaulagaiNo ratings yet
- CoorDocument6 pagesCoordiaaahm58No ratings yet
- Direct Stiffness Method: Direct Stiffness Method: Plane FrameDocument29 pagesDirect Stiffness Method: Direct Stiffness Method: Plane FrameLovneeshNo ratings yet
- Stability of Cable-Arch Structure: Applied Mechanics and Materials October 2012Document8 pagesStability of Cable-Arch Structure: Applied Mechanics and Materials October 2012IbrahimNo ratings yet
- Sulalitha MathsDocument101 pagesSulalitha MathsNithin R Gowda E RNo ratings yet
- Pan Pearl 2021 Paper 2Document14 pagesPan Pearl 2021 Paper 2გიორგი გოგაბერიშვილიNo ratings yet
- Solution of MechanismDocument6 pagesSolution of MechanismMehmet Ali GökkaplanNo ratings yet
- Important Instructions:: Test Booklet CodeDocument27 pagesImportant Instructions:: Test Booklet Codeapi-3721555No ratings yet
- Counting: March 9, 2017Document33 pagesCounting: March 9, 2017Dicky Tak Hin WongNo ratings yet
- Solutions 1Document3 pagesSolutions 1Putu EkaNo ratings yet
- Experiment No. 1 Magnetic Field (Helmholtz Coils)Document6 pagesExperiment No. 1 Magnetic Field (Helmholtz Coils)arihantNo ratings yet
- The Penrose Transform: Its Interaction with Representation TheoryFrom EverandThe Penrose Transform: Its Interaction with Representation TheoryNo ratings yet
- Mathematical Analysis 1: theory and solved exercisesFrom EverandMathematical Analysis 1: theory and solved exercisesRating: 5 out of 5 stars5/5 (1)
- Summary of Hargis Et Al. (2014) CSA HydrationDocument3 pagesSummary of Hargis Et Al. (2014) CSA HydrationSaznizam Sazmee SinohNo ratings yet
- Application of Petrology in Mineral Exploration PDFDocument19 pagesApplication of Petrology in Mineral Exploration PDFPetrology GSITINo ratings yet
- State Mineral Industry MEGHALAYADocument6 pagesState Mineral Industry MEGHALAYAmujib uddin siddiquiNo ratings yet
- Class 12 Set - B: Chemistry - CH 402Document3 pagesClass 12 Set - B: Chemistry - CH 402SonuSharmaNo ratings yet
- The Jade Enigma: Jill HobbsDocument17 pagesThe Jade Enigma: Jill HobbsOuchyNo ratings yet
- Chapter 1: Crystal Structure: Types of SolidsDocument10 pagesChapter 1: Crystal Structure: Types of Solidsnahom teferaNo ratings yet
- Pengaruh Penambahan Unsur Aluminium Murni Pada BahanDocument11 pagesPengaruh Penambahan Unsur Aluminium Murni Pada BahanSiti MaryamNo ratings yet
- MSE304 Assignment IDocument4 pagesMSE304 Assignment IHarshit SachdevNo ratings yet
- X Reserves Resources 201112Document50 pagesX Reserves Resources 201112Eun Young MNo ratings yet
- Math ModelDocument12 pagesMath ModelraparlaskNo ratings yet
- Lab 1: Mineral Identification: Mcgill Epsc 221Document4 pagesLab 1: Mineral Identification: Mcgill Epsc 221Daniel DicksonNo ratings yet
- (Dahle 2001) - Eutectic Nucleation and Growth in Hypoeutectic Al-SI Alloys at Different Strontium LevelsDocument12 pages(Dahle 2001) - Eutectic Nucleation and Growth in Hypoeutectic Al-SI Alloys at Different Strontium Levelsפּואַ פּוגאַNo ratings yet
- Name of Lessee Registration Number Mine - Code Email Id of The Owner/lesseeDocument3 pagesName of Lessee Registration Number Mine - Code Email Id of The Owner/lesseeKapil RampalNo ratings yet
- 1 Basics of X-Ray Powder DiffractionDocument110 pages1 Basics of X-Ray Powder DiffractionAnonymous Waw8C0qkAANo ratings yet
- Norm CalculationDocument5 pagesNorm CalculationmohammedNo ratings yet
- ESCAP 1993 MN Geology Mineral Resources Nepal MapDocument2 pagesESCAP 1993 MN Geology Mineral Resources Nepal MapAditya mahatoNo ratings yet
- GEO101 HW1M1 - Group EDocument20 pagesGEO101 HW1M1 - Group EDE GUIA, ENRICO GABRIEL F.No ratings yet
- MDSW Orissa01Document190 pagesMDSW Orissa01miningnova1No ratings yet
- Crystal LatticeDocument10 pagesCrystal LatticeAmalina NajibNo ratings yet
- Chapter 2 EssayDocument4 pagesChapter 2 EssayarjunNo ratings yet
- Clay Mineral Cements in SandstonesDocument3 pagesClay Mineral Cements in Sandstonesandrea.cipagautaNo ratings yet
- XRD - Karmat 2Document40 pagesXRD - Karmat 2sri ramadhaniNo ratings yet
- High Sulphidation Gold DepositsDocument27 pagesHigh Sulphidation Gold DepositsStanly TandrisNo ratings yet
- Silicon Wafer and Ingot Preparation PDFDocument23 pagesSilicon Wafer and Ingot Preparation PDFprinceNo ratings yet
- Introduction To EBSDDocument16 pagesIntroduction To EBSDNoé SeijoNo ratings yet
- Slides 02Document10 pagesSlides 02Anudwaipaon AntuNo ratings yet
- Topic 03Document19 pagesTopic 03Mohammed AbdoNo ratings yet
- BuildingstoneDocument20 pagesBuildingstoneAlexander AsonkeyNo ratings yet
- All Gems in WorldDocument632 pagesAll Gems in Worldsankar100% (1)
- Solid State Physics 19UPHE02 ODD SEMDocument128 pagesSolid State Physics 19UPHE02 ODD SEMLALITHA MNo ratings yet