
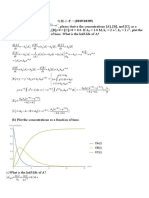
(A) First-Order Reactions Close To Equilibrium: Da Ka K B DT K Ke A A K K
(A) First-Order Reactions Close To Equilibrium: Da Ka K B DT K Ke A A K K
You might also like
- Shared Reading:: The Great EightDocument1 pageShared Reading:: The Great EightursulaNo ratings yet
- Problem Set 5 SolutionDocument17 pagesProblem Set 5 SolutionHarold OngNo ratings yet
- Kinetics Formula SheetDocument1 pageKinetics Formula SheetdfasdfNo ratings yet
- 108 Quiz1Document1 page108 Quiz1沈智恩No ratings yet
- 21.8 (A) Lindermann-Hinshelwood Mechanism: (Slow, RDS) + +Document18 pages21.8 (A) Lindermann-Hinshelwood Mechanism: (Slow, RDS) + +cmc107No ratings yet
- ParallRxn PDFDocument1 pageParallRxn PDFWizra QamarNo ratings yet
- ParallRxn PDFDocument1 pageParallRxn PDFAbhinav RajeshNo ratings yet
- ParallRxn PDFDocument1 pageParallRxn PDFAbhinav RajeshNo ratings yet
- Leibnitz Theorem For NTH DerivativeDocument5 pagesLeibnitz Theorem For NTH Derivativevijay kumar honnaliNo ratings yet
- 09PM I SolDocument8 pages09PM I Solapi-25887708No ratings yet
- 09PM - Full SolDocument17 pages09PM - Full Solapi-25887708No ratings yet
- Integral de LebesgueDocument6 pagesIntegral de LebesgueEnrique PradoNo ratings yet
- Laplace AnaliticaDocument2 pagesLaplace AnaliticaValaur Ekbalam M BNo ratings yet
- Lecture3 (Adsorption-Part2) 2022Document8 pagesLecture3 (Adsorption-Part2) 2022최종윤No ratings yet
- Exercise 2.3: Part (A)Document3 pagesExercise 2.3: Part (A)Miguel SantanaNo ratings yet
- Answers 2 BDocument11 pagesAnswers 2 BMarc Samper SeguraNo ratings yet
- Assignment - 2 Sol PDFDocument7 pagesAssignment - 2 Sol PDFGreco S50No ratings yet
- 11PM Sol FullDocument18 pages11PM Sol FullKam Pang LeungNo ratings yet
- Desafio 3 - Infer Encia Estat Istica: 1 Resolu C AoDocument5 pagesDesafio 3 - Infer Encia Estat Istica: 1 Resolu C AoJuliano HerzogNo ratings yet
- Application of Partial Fraction in Chemical Kinetics and PharmacokineticsDocument9 pagesApplication of Partial Fraction in Chemical Kinetics and PharmacokineticsAshish MauryaNo ratings yet
- MMSB Exercise SolutionsDocument49 pagesMMSB Exercise SolutionsYolanda Winarny Eviphanie HutabaratNo ratings yet
- Linear ReportDocument3 pagesLinear ReportLuffy D. BrownsNo ratings yet
- Reglas de IntegraciónDocument1 pageReglas de IntegraciónAndres RangelNo ratings yet
- EE1101: Signals and Systems Tutorial 3 SolutionsDocument19 pagesEE1101: Signals and Systems Tutorial 3 SolutionsRohan Kumar ee21b113No ratings yet
- Real Business Cycles: Jes Us Fern Andez-Villaverde University of PennsylvaniaDocument22 pagesReal Business Cycles: Jes Us Fern Andez-Villaverde University of PennsylvaniawilianssterNo ratings yet
- 2024 05 06 18 31 SolutionDocument40 pages2024 05 06 18 31 SolutionGANIT WALANo ratings yet
- Part-02 Reaction Kinetics (KINETIKA KIMIA, 15 DES 2020)Document24 pagesPart-02 Reaction Kinetics (KINETIKA KIMIA, 15 DES 2020)Anisa NurulNo ratings yet
- Ps8sol 6245 2004Document6 pagesPs8sol 6245 2004Tejas PatilNo ratings yet
- Tabla de PrimitivasDocument1 pageTabla de PrimitivasaullidodealmaNo ratings yet
- Manual CRQ U3 2022 Deduccion FormulasDocument21 pagesManual CRQ U3 2022 Deduccion FormulasOliver Alex MoralesNo ratings yet
- 08PM I SolDocument10 pages08PM I Solapi-25887708100% (1)
- Chapter 14. Enzyme KineticsDocument44 pagesChapter 14. Enzyme KineticsKalai VaniNo ratings yet
- Problem Set 1Document4 pagesProblem Set 1حيدر علاء عبدالزهره جاسمNo ratings yet
- Persamaan Co StateDocument1 pagePersamaan Co Stateshofiya dwiNo ratings yet
- K XK XK X K XK Yk Yk Ykn Ykn: 7.9 State-Space Realizations 7.9.a Controllable Canonical RealizationDocument9 pagesK XK XK X K XK Yk Yk Ykn Ykn: 7.9 State-Space Realizations 7.9.a Controllable Canonical RealizationSayali LomateNo ratings yet
- Basic Matrix Operations: - TransposeDocument26 pagesBasic Matrix Operations: - Transposesaraei7142356No ratings yet
- EE402 Lecture 3Document5 pagesEE402 Lecture 3sdfgNo ratings yet
- CHM172 The Rates of Reactions - Integrated Rate LawsDocument26 pagesCHM172 The Rates of Reactions - Integrated Rate LawsGezem GigantoNo ratings yet
- Avance Fundamental 4 CODocument6 pagesAvance Fundamental 4 COSerna ReynaNo ratings yet
- Rumus Dan Persamaan Yang Dapat Digunakan (Soal Farmakokinetika)Document1 pageRumus Dan Persamaan Yang Dapat Digunakan (Soal Farmakokinetika)asriNo ratings yet
- Documento 1Document4 pagesDocumento 1Ing. Santos BlackNo ratings yet
- 1theorem-3 1 6-To-Theorem-3 1 10 PDFDocument97 pages1theorem-3 1 6-To-Theorem-3 1 10 PDFRonelo Amancio FajardoNo ratings yet
- Course Reader Spring 2014Document23 pagesCourse Reader Spring 2014sebastian nasiNo ratings yet
- Chemical OscillationsDocument4 pagesChemical OscillationssouryadeepmitraNo ratings yet
- 10Document13 pages10cmc107No ratings yet
- Macro Theory110 Assignment 5Document8 pagesMacro Theory110 Assignment 5Greco S50No ratings yet
- Solving Real Business Cycle Models by Solving Systems of First Order ConditionsDocument18 pagesSolving Real Business Cycle Models by Solving Systems of First Order ConditionsBrunéNo ratings yet
- Final SolutionsDocument14 pagesFinal SolutionsZainal AbidinNo ratings yet
- 1 Lecture Notes: The Solow ModelDocument5 pages1 Lecture Notes: The Solow Modelmrs dosadoNo ratings yet
- Homework 4 SolutionDocument2 pagesHomework 4 SolutionJackNgNo ratings yet
- Fundamental Concepts: Vectors: AB I J JK I JKDocument11 pagesFundamental Concepts: Vectors: AB I J JK I JKmrmodanoNo ratings yet
- DM - Dec 2016Document11 pagesDM - Dec 2016Aditya DumbreNo ratings yet
- Pmath p1q02Document4 pagesPmath p1q02api-19979554No ratings yet
- Formulas For Control System Design: State SpaceDocument3 pagesFormulas For Control System Design: State SpacehanythekingNo ratings yet
- Mathematical Relations PDFDocument229 pagesMathematical Relations PDFmars100% (1)
- Homework5 SolDocument6 pagesHomework5 SolMd Nur-A-Adam DonyNo ratings yet
- T1A/T1B, Hmcheungae Math 2121 Tutorial 4 October 4, 2018Document6 pagesT1A/T1B, Hmcheungae Math 2121 Tutorial 4 October 4, 2018Toby ChengNo ratings yet
- 12 Mathematics Ncert ch03 Matrices Misc SolDocument6 pages12 Mathematics Ncert ch03 Matrices Misc SolChetnaNo ratings yet
- Theorem 3.6: (Binomial Theorem For A Positive Integral Index) Statement: For Any Natural Number NDocument2 pagesTheorem 3.6: (Binomial Theorem For A Positive Integral Index) Statement: For Any Natural Number Ns_amalorpavarajNo ratings yet
- De Moiver's Theorem (Trigonometry) Mathematics Question BankFrom EverandDe Moiver's Theorem (Trigonometry) Mathematics Question BankNo ratings yet
- 10Document13 pages10cmc107No ratings yet
- Equilibrium - Structure - Change: Thermodynamics Quantum TheoryDocument20 pagesEquilibrium - Structure - Change: Thermodynamics Quantum Theorycmc107No ratings yet
- Manho LimDocument25 pagesManho Limcmc107No ratings yet
- McQuarrie Simon Physical Chemistry Solutions 220419 183845Document26 pagesMcQuarrie Simon Physical Chemistry Solutions 220419 183845cmc107No ratings yet
- 4 +Proline-Catalyzed+AsymmetriDocument8 pages4 +Proline-Catalyzed+Asymmetricmc107No ratings yet
- 8 +Peptide+SynthesisDocument7 pages8 +Peptide+Synthesiscmc107No ratings yet
- 1 +Nitraion+of+Methyl+BenzoatePDF 220917 151603Document4 pages1 +Nitraion+of+Methyl+BenzoatePDF 220917 151603cmc107No ratings yet
- 5 +crystallization +purificatiDocument11 pages5 +crystallization +purificaticmc107No ratings yet
- The Enlightenment, The Age of ReasonDocument40 pagesThe Enlightenment, The Age of ReasonRahma MohamedNo ratings yet
- Controller FIS134 19-22 EN PDFDocument4 pagesController FIS134 19-22 EN PDFrimce77No ratings yet
- Material Inspection Request - CIVIL - QF - R-MIR-C-Q-281 (23 - 10 - 2023)Document4 pagesMaterial Inspection Request - CIVIL - QF - R-MIR-C-Q-281 (23 - 10 - 2023)krishanNo ratings yet
- +++ James Arthur Ray - The Science of Success +++Document92 pages+++ James Arthur Ray - The Science of Success +++simona_simona_1982100% (4)
- 2 6 2 English L - LDocument16 pages2 6 2 English L - LAyush RajNo ratings yet
- TCGI - ESG Diploma BrochureDocument11 pagesTCGI - ESG Diploma BrochureIshtiaque AhmedNo ratings yet
- Fenwick - The Failure of The League of NationsDocument5 pagesFenwick - The Failure of The League of NationsEmre CinarNo ratings yet
- LC Vco ThesisDocument8 pagesLC Vco Thesissarareedannarbor100% (2)
- Suzlon One EarthDocument1 pageSuzlon One EarthShubhamNo ratings yet
- Department of Computer EngineeringDocument79 pagesDepartment of Computer EngineeringTejaswi SuryaNo ratings yet
- Hydraulic Fluid Purpose & Properties: (Chapter 2)Document29 pagesHydraulic Fluid Purpose & Properties: (Chapter 2)christodoulos charalambousNo ratings yet
- The Awesome Classroom Design CommitteeDocument3 pagesThe Awesome Classroom Design CommitteeVatamanu ValeriaNo ratings yet
- EPP E2 Activity 2Document2 pagesEPP E2 Activity 2Hannah Nore TalingtingNo ratings yet
- Lesson Plan Observation Grade 12 PhysicsDocument4 pagesLesson Plan Observation Grade 12 PhysicsGerald BaculnaNo ratings yet
- 1lmd Eng 1Document15 pages1lmd Eng 1Yassine HamdiaNo ratings yet
- DGR-7 7 A-OutlineDocument1 pageDGR-7 7 A-OutlineSHERIEFNo ratings yet
- AISI 1045 cs44Document2 pagesAISI 1045 cs44Russell ShacklefordNo ratings yet
- WDM11 01 Que 20210304Document32 pagesWDM11 01 Que 20210304윤소리No ratings yet
- ELA Syllabus Grade 2Document19 pagesELA Syllabus Grade 2Farzam GarshasbiNo ratings yet
- Sidiq Subandriyanto Setyawan: Statement of ParticipationDocument2 pagesSidiq Subandriyanto Setyawan: Statement of Participationsetyawan punkNo ratings yet
- FIFA 19 Squad Builder - FUTBINDocument2 pagesFIFA 19 Squad Builder - FUTBINMauro Vázquez RebolloNo ratings yet
- 30 Must Read Bestsellers in Infographics by HeadwayDocument69 pages30 Must Read Bestsellers in Infographics by HeadwaykateNo ratings yet
- Tutorial 2 EOPDocument3 pagesTutorial 2 EOPammarNo ratings yet
- An Analysis of Swearing Word Types and Translation Techniques in Shaft Movie SubtitlesDocument16 pagesAn Analysis of Swearing Word Types and Translation Techniques in Shaft Movie SubtitlesSoraNo ratings yet
- 10.1016@j.jsames.2017.02.009 4Document17 pages10.1016@j.jsames.2017.02.009 4dkurniadiNo ratings yet
- Hardware Manual of The Easy Servo Drives: ES-DH SeriesDocument28 pagesHardware Manual of The Easy Servo Drives: ES-DH Serieshuutan12345No ratings yet
- About The Job Smelter Operation ReadinessDocument4 pagesAbout The Job Smelter Operation Readinessmuhammad ridwanNo ratings yet
- James Dobson HomeworkDocument6 pagesJames Dobson Homeworkafmsougjd100% (1)
- 450-Article Text-1408-1-10-20210324Document8 pages450-Article Text-1408-1-10-20210324noitoloh24No ratings yet































































































Download as pdf or txt
You might also like
- Shared Reading:: The Great EightDocument1 pageShared Reading:: The Great EightursulaNo ratings yet
- Problem Set 5 SolutionDocument17 pagesProblem Set 5 SolutionHarold OngNo ratings yet
- Kinetics Formula SheetDocument1 pageKinetics Formula SheetdfasdfNo ratings yet
- 108 Quiz1Document1 page108 Quiz1沈智恩No ratings yet
- 21.8 (A) Lindermann-Hinshelwood Mechanism: (Slow, RDS) + +Document18 pages21.8 (A) Lindermann-Hinshelwood Mechanism: (Slow, RDS) + +cmc107No ratings yet
- ParallRxn PDFDocument1 pageParallRxn PDFWizra QamarNo ratings yet
- ParallRxn PDFDocument1 pageParallRxn PDFAbhinav RajeshNo ratings yet
- ParallRxn PDFDocument1 pageParallRxn PDFAbhinav RajeshNo ratings yet
- Leibnitz Theorem For NTH DerivativeDocument5 pagesLeibnitz Theorem For NTH Derivativevijay kumar honnaliNo ratings yet
- 09PM I SolDocument8 pages09PM I Solapi-25887708No ratings yet
- 09PM - Full SolDocument17 pages09PM - Full Solapi-25887708No ratings yet
- Integral de LebesgueDocument6 pagesIntegral de LebesgueEnrique PradoNo ratings yet
- Laplace AnaliticaDocument2 pagesLaplace AnaliticaValaur Ekbalam M BNo ratings yet
- Lecture3 (Adsorption-Part2) 2022Document8 pagesLecture3 (Adsorption-Part2) 2022최종윤No ratings yet
- Exercise 2.3: Part (A)Document3 pagesExercise 2.3: Part (A)Miguel SantanaNo ratings yet
- Answers 2 BDocument11 pagesAnswers 2 BMarc Samper SeguraNo ratings yet
- Assignment - 2 Sol PDFDocument7 pagesAssignment - 2 Sol PDFGreco S50No ratings yet
- 11PM Sol FullDocument18 pages11PM Sol FullKam Pang LeungNo ratings yet
- Desafio 3 - Infer Encia Estat Istica: 1 Resolu C AoDocument5 pagesDesafio 3 - Infer Encia Estat Istica: 1 Resolu C AoJuliano HerzogNo ratings yet
- Application of Partial Fraction in Chemical Kinetics and PharmacokineticsDocument9 pagesApplication of Partial Fraction in Chemical Kinetics and PharmacokineticsAshish MauryaNo ratings yet
- MMSB Exercise SolutionsDocument49 pagesMMSB Exercise SolutionsYolanda Winarny Eviphanie HutabaratNo ratings yet
- Linear ReportDocument3 pagesLinear ReportLuffy D. BrownsNo ratings yet
- Reglas de IntegraciónDocument1 pageReglas de IntegraciónAndres RangelNo ratings yet
- EE1101: Signals and Systems Tutorial 3 SolutionsDocument19 pagesEE1101: Signals and Systems Tutorial 3 SolutionsRohan Kumar ee21b113No ratings yet
- Real Business Cycles: Jes Us Fern Andez-Villaverde University of PennsylvaniaDocument22 pagesReal Business Cycles: Jes Us Fern Andez-Villaverde University of PennsylvaniawilianssterNo ratings yet
- 2024 05 06 18 31 SolutionDocument40 pages2024 05 06 18 31 SolutionGANIT WALANo ratings yet
- Part-02 Reaction Kinetics (KINETIKA KIMIA, 15 DES 2020)Document24 pagesPart-02 Reaction Kinetics (KINETIKA KIMIA, 15 DES 2020)Anisa NurulNo ratings yet
- Ps8sol 6245 2004Document6 pagesPs8sol 6245 2004Tejas PatilNo ratings yet
- Tabla de PrimitivasDocument1 pageTabla de PrimitivasaullidodealmaNo ratings yet
- Manual CRQ U3 2022 Deduccion FormulasDocument21 pagesManual CRQ U3 2022 Deduccion FormulasOliver Alex MoralesNo ratings yet
- 08PM I SolDocument10 pages08PM I Solapi-25887708100% (1)
- Chapter 14. Enzyme KineticsDocument44 pagesChapter 14. Enzyme KineticsKalai VaniNo ratings yet
- Problem Set 1Document4 pagesProblem Set 1حيدر علاء عبدالزهره جاسمNo ratings yet
- Persamaan Co StateDocument1 pagePersamaan Co Stateshofiya dwiNo ratings yet
- K XK XK X K XK Yk Yk Ykn Ykn: 7.9 State-Space Realizations 7.9.a Controllable Canonical RealizationDocument9 pagesK XK XK X K XK Yk Yk Ykn Ykn: 7.9 State-Space Realizations 7.9.a Controllable Canonical RealizationSayali LomateNo ratings yet
- Basic Matrix Operations: - TransposeDocument26 pagesBasic Matrix Operations: - Transposesaraei7142356No ratings yet
- EE402 Lecture 3Document5 pagesEE402 Lecture 3sdfgNo ratings yet
- CHM172 The Rates of Reactions - Integrated Rate LawsDocument26 pagesCHM172 The Rates of Reactions - Integrated Rate LawsGezem GigantoNo ratings yet
- Avance Fundamental 4 CODocument6 pagesAvance Fundamental 4 COSerna ReynaNo ratings yet
- Rumus Dan Persamaan Yang Dapat Digunakan (Soal Farmakokinetika)Document1 pageRumus Dan Persamaan Yang Dapat Digunakan (Soal Farmakokinetika)asriNo ratings yet
- Documento 1Document4 pagesDocumento 1Ing. Santos BlackNo ratings yet
- 1theorem-3 1 6-To-Theorem-3 1 10 PDFDocument97 pages1theorem-3 1 6-To-Theorem-3 1 10 PDFRonelo Amancio FajardoNo ratings yet
- Course Reader Spring 2014Document23 pagesCourse Reader Spring 2014sebastian nasiNo ratings yet
- Chemical OscillationsDocument4 pagesChemical OscillationssouryadeepmitraNo ratings yet
- 10Document13 pages10cmc107No ratings yet
- Macro Theory110 Assignment 5Document8 pagesMacro Theory110 Assignment 5Greco S50No ratings yet
- Solving Real Business Cycle Models by Solving Systems of First Order ConditionsDocument18 pagesSolving Real Business Cycle Models by Solving Systems of First Order ConditionsBrunéNo ratings yet
- Final SolutionsDocument14 pagesFinal SolutionsZainal AbidinNo ratings yet
- 1 Lecture Notes: The Solow ModelDocument5 pages1 Lecture Notes: The Solow Modelmrs dosadoNo ratings yet
- Homework 4 SolutionDocument2 pagesHomework 4 SolutionJackNgNo ratings yet
- Fundamental Concepts: Vectors: AB I J JK I JKDocument11 pagesFundamental Concepts: Vectors: AB I J JK I JKmrmodanoNo ratings yet
- DM - Dec 2016Document11 pagesDM - Dec 2016Aditya DumbreNo ratings yet
- Pmath p1q02Document4 pagesPmath p1q02api-19979554No ratings yet
- Formulas For Control System Design: State SpaceDocument3 pagesFormulas For Control System Design: State SpacehanythekingNo ratings yet
- Mathematical Relations PDFDocument229 pagesMathematical Relations PDFmars100% (1)
- Homework5 SolDocument6 pagesHomework5 SolMd Nur-A-Adam DonyNo ratings yet
- T1A/T1B, Hmcheungae Math 2121 Tutorial 4 October 4, 2018Document6 pagesT1A/T1B, Hmcheungae Math 2121 Tutorial 4 October 4, 2018Toby ChengNo ratings yet
- 12 Mathematics Ncert ch03 Matrices Misc SolDocument6 pages12 Mathematics Ncert ch03 Matrices Misc SolChetnaNo ratings yet
- Theorem 3.6: (Binomial Theorem For A Positive Integral Index) Statement: For Any Natural Number NDocument2 pagesTheorem 3.6: (Binomial Theorem For A Positive Integral Index) Statement: For Any Natural Number Ns_amalorpavarajNo ratings yet
- De Moiver's Theorem (Trigonometry) Mathematics Question BankFrom EverandDe Moiver's Theorem (Trigonometry) Mathematics Question BankNo ratings yet
- 10Document13 pages10cmc107No ratings yet
- Equilibrium - Structure - Change: Thermodynamics Quantum TheoryDocument20 pagesEquilibrium - Structure - Change: Thermodynamics Quantum Theorycmc107No ratings yet
- Manho LimDocument25 pagesManho Limcmc107No ratings yet
- McQuarrie Simon Physical Chemistry Solutions 220419 183845Document26 pagesMcQuarrie Simon Physical Chemistry Solutions 220419 183845cmc107No ratings yet
- 4 +Proline-Catalyzed+AsymmetriDocument8 pages4 +Proline-Catalyzed+Asymmetricmc107No ratings yet
- 8 +Peptide+SynthesisDocument7 pages8 +Peptide+Synthesiscmc107No ratings yet
- 1 +Nitraion+of+Methyl+BenzoatePDF 220917 151603Document4 pages1 +Nitraion+of+Methyl+BenzoatePDF 220917 151603cmc107No ratings yet
- 5 +crystallization +purificatiDocument11 pages5 +crystallization +purificaticmc107No ratings yet
- The Enlightenment, The Age of ReasonDocument40 pagesThe Enlightenment, The Age of ReasonRahma MohamedNo ratings yet
- Controller FIS134 19-22 EN PDFDocument4 pagesController FIS134 19-22 EN PDFrimce77No ratings yet
- Material Inspection Request - CIVIL - QF - R-MIR-C-Q-281 (23 - 10 - 2023)Document4 pagesMaterial Inspection Request - CIVIL - QF - R-MIR-C-Q-281 (23 - 10 - 2023)krishanNo ratings yet
- +++ James Arthur Ray - The Science of Success +++Document92 pages+++ James Arthur Ray - The Science of Success +++simona_simona_1982100% (4)
- 2 6 2 English L - LDocument16 pages2 6 2 English L - LAyush RajNo ratings yet
- TCGI - ESG Diploma BrochureDocument11 pagesTCGI - ESG Diploma BrochureIshtiaque AhmedNo ratings yet
- Fenwick - The Failure of The League of NationsDocument5 pagesFenwick - The Failure of The League of NationsEmre CinarNo ratings yet
- LC Vco ThesisDocument8 pagesLC Vco Thesissarareedannarbor100% (2)
- Suzlon One EarthDocument1 pageSuzlon One EarthShubhamNo ratings yet
- Department of Computer EngineeringDocument79 pagesDepartment of Computer EngineeringTejaswi SuryaNo ratings yet
- Hydraulic Fluid Purpose & Properties: (Chapter 2)Document29 pagesHydraulic Fluid Purpose & Properties: (Chapter 2)christodoulos charalambousNo ratings yet
- The Awesome Classroom Design CommitteeDocument3 pagesThe Awesome Classroom Design CommitteeVatamanu ValeriaNo ratings yet
- EPP E2 Activity 2Document2 pagesEPP E2 Activity 2Hannah Nore TalingtingNo ratings yet
- Lesson Plan Observation Grade 12 PhysicsDocument4 pagesLesson Plan Observation Grade 12 PhysicsGerald BaculnaNo ratings yet
- 1lmd Eng 1Document15 pages1lmd Eng 1Yassine HamdiaNo ratings yet
- DGR-7 7 A-OutlineDocument1 pageDGR-7 7 A-OutlineSHERIEFNo ratings yet
- AISI 1045 cs44Document2 pagesAISI 1045 cs44Russell ShacklefordNo ratings yet
- WDM11 01 Que 20210304Document32 pagesWDM11 01 Que 20210304윤소리No ratings yet
- ELA Syllabus Grade 2Document19 pagesELA Syllabus Grade 2Farzam GarshasbiNo ratings yet
- Sidiq Subandriyanto Setyawan: Statement of ParticipationDocument2 pagesSidiq Subandriyanto Setyawan: Statement of Participationsetyawan punkNo ratings yet
- FIFA 19 Squad Builder - FUTBINDocument2 pagesFIFA 19 Squad Builder - FUTBINMauro Vázquez RebolloNo ratings yet
- 30 Must Read Bestsellers in Infographics by HeadwayDocument69 pages30 Must Read Bestsellers in Infographics by HeadwaykateNo ratings yet
- Tutorial 2 EOPDocument3 pagesTutorial 2 EOPammarNo ratings yet
- An Analysis of Swearing Word Types and Translation Techniques in Shaft Movie SubtitlesDocument16 pagesAn Analysis of Swearing Word Types and Translation Techniques in Shaft Movie SubtitlesSoraNo ratings yet
- 10.1016@j.jsames.2017.02.009 4Document17 pages10.1016@j.jsames.2017.02.009 4dkurniadiNo ratings yet
- Hardware Manual of The Easy Servo Drives: ES-DH SeriesDocument28 pagesHardware Manual of The Easy Servo Drives: ES-DH Serieshuutan12345No ratings yet
- About The Job Smelter Operation ReadinessDocument4 pagesAbout The Job Smelter Operation Readinessmuhammad ridwanNo ratings yet
- James Dobson HomeworkDocument6 pagesJames Dobson Homeworkafmsougjd100% (1)
- 450-Article Text-1408-1-10-20210324Document8 pages450-Article Text-1408-1-10-20210324noitoloh24No ratings yet