Download as pdf or txt
You might also like
- Ch.24.1 Intro To AnimalsDocument20 pagesCh.24.1 Intro To AnimalsweldeenytNo ratings yet
- Road To The Moon - A Great Occult Story - DR George W CareyDocument30 pagesRoad To The Moon - A Great Occult Story - DR George W CareyBret Trismegistus100% (4)
- Disorders of Growth, Differentiation and Morphogenesis EditDocument48 pagesDisorders of Growth, Differentiation and Morphogenesis EditRizka Desti AyuniNo ratings yet
- US Army Survival Manual FM 21-76 1992Document648 pagesUS Army Survival Manual FM 21-76 1992Total Prepper100% (6)
- 2020-Nanopore Basecalling From A Perspective of Instance SegmentationDocument9 pages2020-Nanopore Basecalling From A Perspective of Instance SegmentationMichael LeeNo ratings yet
- A World of Opportunities With Nanopore SequencingDocument11 pagesA World of Opportunities With Nanopore SequencingShavana RajkumarNo ratings yet
- Nanopore Sequencing Review of Potential Applications inDocument11 pagesNanopore Sequencing Review of Potential Applications inMohamed ChahmiNo ratings yet
- Senol Cali Et Al., 2018Document18 pagesSenol Cali Et Al., 2018Javier VázquezNo ratings yet
- Nanopore SequencingDocument16 pagesNanopore SequencingJohnNo ratings yet
- Minion-A Pocket Sized Wonder: By, Sutrishna Ghosh Anwesha Bose Ananya ChakrabortyDocument13 pagesMinion-A Pocket Sized Wonder: By, Sutrishna Ghosh Anwesha Bose Ananya ChakrabortyAnwesha BoseNo ratings yet
- Performance of Neural Network Basecalling Tools For Oxford Nanopore SequencingDocument10 pagesPerformance of Neural Network Basecalling Tools For Oxford Nanopore SequencingMichael LeeNo ratings yet
- Biosensors: Nanopore Technology and Its Applications in Gene SequencingDocument17 pagesBiosensors: Nanopore Technology and Its Applications in Gene Sequencingandres barreraNo ratings yet
- Miranalyzer: A Microrna Detection and Analysis Tool For Next-Generation Sequencing ExperimentsDocument9 pagesMiranalyzer: A Microrna Detection and Analysis Tool For Next-Generation Sequencing ExperimentsFrontiersNo ratings yet
- Nanopore Sensing 5733 WACwTEsDocument15 pagesNanopore Sensing 5733 WACwTEsSahana CNo ratings yet
- An Overview of SequencingHigh Throughput SequencingDocument11 pagesAn Overview of SequencingHigh Throughput SequencingAnh NguyenNo ratings yet
- Comparison of High Throughput Next GenerDocument12 pagesComparison of High Throughput Next GenerAni IoanaNo ratings yet
- Miranalyzer: A Microrna Detection and Analysis Tool For Next-Generation Sequencing ExperimentsDocument9 pagesMiranalyzer: A Microrna Detection and Analysis Tool For Next-Generation Sequencing ExperimentsJorge Hantar Touma LazoNo ratings yet
- Oxford Nanopore Minion Sequencing and Genome Assembly: Genomics Proteomics BioinformaticsDocument15 pagesOxford Nanopore Minion Sequencing and Genome Assembly: Genomics Proteomics BioinformaticsSadio KeitaNo ratings yet
- Bioinformatics Approaches For Genomics ADocument17 pagesBioinformatics Approaches For Genomics AZay Yar WinNo ratings yet
- Opportunities and Challenges in Long-Read Sequencing Data AnalysisDocument16 pagesOpportunities and Challenges in Long-Read Sequencing Data AnalysisSuran Ravindran NambisanNo ratings yet
- Bioinformatics New Tools and Applications in LifeDocument16 pagesBioinformatics New Tools and Applications in LifedffNo ratings yet
- Unlocking The Power of NanoporesDocument14 pagesUnlocking The Power of NanoporesRiyaNo ratings yet
- Comparion of Polishing ToolsDocument11 pagesComparion of Polishing ToolsAdwikaNo ratings yet
- 10.1007@s12275 020 9556 yDocument13 pages10.1007@s12275 020 9556 yClaudia MaturanaNo ratings yet
- Porreca2010 PDFDocument2 pagesPorreca2010 PDFBeatriz De Vicente MartinezNo ratings yet
- High-Throughput Sequencing Technology and Its Application: SciencedirectDocument13 pagesHigh-Throughput Sequencing Technology and Its Application: SciencedirectamarNo ratings yet
- HPDTRP 2008 10 Su WangDocument9 pagesHPDTRP 2008 10 Su WangDonna A. GarciaNo ratings yet
- CA 2 Nissey Ma'amDocument5 pagesCA 2 Nissey Ma'amLakshya PhoolwaniNo ratings yet
- Next Generation SequencingDocument7 pagesNext Generation SequencingAvani KaushalNo ratings yet
- Equipe1 - A Tale of Three Next Generation Sequencing Platforms - Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq SequencersDocument13 pagesEquipe1 - A Tale of Three Next Generation Sequencing Platforms - Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq SequencersFagnerOliveiraNo ratings yet
- Sequencing Technologies - The Next Generation: Michael L. MetzkerDocument16 pagesSequencing Technologies - The Next Generation: Michael L. MetzkerWayne JiaoNo ratings yet
- Base-Calling For Next-Generation Sequencing Platforms: Briefings in Bioinformatics September 2011Document10 pagesBase-Calling For Next-Generation Sequencing Platforms: Briefings in Bioinformatics September 2011karen CJNo ratings yet
- Zhang 2019 IOP Conf. Ser. Earth Environ. Sci. 332 042003Document7 pagesZhang 2019 IOP Conf. Ser. Earth Environ. Sci. 332 042003Sujitha maryNo ratings yet
- SMC2018 ReviewDocument7 pagesSMC2018 Reviewcharliee8001No ratings yet
- Nano RoboticsDocument13 pagesNano RoboticsKrithika ChandrasekaranNo ratings yet
- A Scalable Hardware Accelerator For Mobile DNA SequencingDocument14 pagesA Scalable Hardware Accelerator For Mobile DNA Sequencingroopa_kothapalliNo ratings yet
- Next Generation Sequencing Platforms PDFDocument5 pagesNext Generation Sequencing Platforms PDFvedanidhiNo ratings yet
- Oxford Nanopore Product Brochure Sept2023Document19 pagesOxford Nanopore Product Brochure Sept2023Devansh PanwarNo ratings yet
- 100 Gbitss Integrated Quantum Random Number Generator Based On Vacuum FluctuationsDocument12 pages100 Gbitss Integrated Quantum Random Number Generator Based On Vacuum Fluctuationsali hadi amhazNo ratings yet
- LAb On A Chip TechnologyDocument10 pagesLAb On A Chip TechnologyAbhishek GuptaNo ratings yet
- Trapnell 2009Document7 pagesTrapnell 2009László SágiNo ratings yet
- Next Generation Sequencing and Sequence Assembly: Methodologies and AlgorithmsFrom EverandNext Generation Sequencing and Sequence Assembly: Methodologies and AlgorithmsNo ratings yet
- Overview of Next Generation Sequencing TechnologiesDocument12 pagesOverview of Next Generation Sequencing TechnologiesiuventasNo ratings yet
- Nanorobots in Medicine-A New Dimension in Bio NanotechnologyDocument6 pagesNanorobots in Medicine-A New Dimension in Bio NanotechnologyLeonardo GuerraNo ratings yet
- DNA Nanotechnology Assisted Nanopore-BasedDocument16 pagesDNA Nanotechnology Assisted Nanopore-BasedIsabela DragomirNo ratings yet
- Metodi Bioinformatici Per L'analisi Del Genoma UmanoDocument148 pagesMetodi Bioinformatici Per L'analisi Del Genoma Umanoparetini01No ratings yet
- Ngs Coronavirus App Note 1270 2020 001Document4 pagesNgs Coronavirus App Note 1270 2020 001sylvi293No ratings yet
- Wireless Paper-Based Biosensor Reader For The Detection of Infectious Diseases at The Point of CareDocument3 pagesWireless Paper-Based Biosensor Reader For The Detection of Infectious Diseases at The Point of Caresanthutammineni3No ratings yet
- Targeted Nanopore Sequencing by Real-Time Mapping of Raw ElectricalDocument36 pagesTargeted Nanopore Sequencing by Real-Time Mapping of Raw ElectricalMichael LeeNo ratings yet
- Bioinformatics: Applications NoteDocument2 pagesBioinformatics: Applications NoteRicardo GarcíaNo ratings yet
- Introduction To Sequencing and ONT DR Linzy EltonDocument32 pagesIntroduction To Sequencing and ONT DR Linzy EltongiangdpNo ratings yet
- Simulaiton ModelDocument18 pagesSimulaiton ModelnomanNo ratings yet
- Anandhu ReportDocument23 pagesAnandhu ReportSarin Sayal100% (1)
- Review of Massively Parallel DNA Sequencing TechnoDocument13 pagesReview of Massively Parallel DNA Sequencing TechnoLamia FahadNo ratings yet
- Detecting The DNA of Dengue Serotype 2 Using Aluminium Nanoparticle Doped Zinc Oxide Nanostructure: Synthesis, Analysis and CharacterizationDocument9 pagesDetecting The DNA of Dengue Serotype 2 Using Aluminium Nanoparticle Doped Zinc Oxide Nanostructure: Synthesis, Analysis and CharacterizationGayatree TripathyNo ratings yet
- Baloglu 2021 A Workflow For Accurate MetabarcodiDocument11 pagesBaloglu 2021 A Workflow For Accurate MetabarcodiHana MikoNo ratings yet
- Next GenerationDocument5 pagesNext GenerationWajiha FatimaNo ratings yet
- Paper 2Document10 pagesPaper 2MalikNo ratings yet
- Advance Sequencing TechnologyDocument24 pagesAdvance Sequencing TechnologyvinothjayamaniNo ratings yet
- Unit-14 NANOBIOANALYTICAL TECHNIQUESDocument14 pagesUnit-14 NANOBIOANALYTICAL TECHNIQUESAhmad AliNo ratings yet
- Metagenomic Shotgun Seq Learning ProgressDocument19 pagesMetagenomic Shotgun Seq Learning ProgressAnisaLFNo ratings yet
- SmartFlare RNA Detection ProbeDocument73 pagesSmartFlare RNA Detection ProbeFatma M. Barakat100% (1)
- Nanoj: A High-Performance Open-Source Super-Resolution Microscopy ToolboxDocument11 pagesNanoj: A High-Performance Open-Source Super-Resolution Microscopy ToolboxDp PdNo ratings yet
- NanoRobotics DOCUMENTDocument16 pagesNanoRobotics DOCUMENTAnirudh D DyagaNo ratings yet
- Protocol Experimental 2009Document13 pagesProtocol Experimental 2009Iuliana SoldanescuNo ratings yet
- 10.1016@b978 0 12 409547 2.13307 4Document10 pages10.1016@b978 0 12 409547 2.13307 4Iuliana SoldanescuNo ratings yet
- NewimageDocument1 pageNewimageIuliana SoldanescuNo ratings yet
- Miller2017 ReviewDocument48 pagesMiller2017 ReviewIuliana SoldanescuNo ratings yet
- Controlling Molecular Transport Through Nanopores: Ulrich F. KeyserDocument10 pagesControlling Molecular Transport Through Nanopores: Ulrich F. KeyserIuliana SoldanescuNo ratings yet
- 10 1002@smtd 201900595luchianDocument13 pages10 1002@smtd 201900595luchianIuliana SoldanescuNo ratings yet
- Molecular Electronics Devices A Short Review - Delnero2010Document14 pagesMolecular Electronics Devices A Short Review - Delnero2010Iuliana SoldanescuNo ratings yet
- Song 1996Document8 pagesSong 1996Iuliana SoldanescuNo ratings yet
- Single Molecule Sensing by Nanopores and Nanopore Devices: Li-Qun Gu and Ji Wook ShimDocument11 pagesSingle Molecule Sensing by Nanopores and Nanopore Devices: Li-Qun Gu and Ji Wook ShimIuliana SoldanescuNo ratings yet
- Braha 1999Document5 pagesBraha 1999Iuliana SoldanescuNo ratings yet
- Uram2008 NoiseDocument16 pagesUram2008 NoiseIuliana SoldanescuNo ratings yet
- UNIT 6 Student Handout2023-IDocument12 pagesUNIT 6 Student Handout2023-IPaola Mercedes Matos OrtegaNo ratings yet
- Tensile Test of Copper Fibers in Conformance With ASTM C1557 Using Agilent UTM T150Document4 pagesTensile Test of Copper Fibers in Conformance With ASTM C1557 Using Agilent UTM T150Celeste LopezNo ratings yet
- Bamboo Species Source List No. 36 2017-2018Document56 pagesBamboo Species Source List No. 36 2017-2018André MichaelNo ratings yet
- 1st Midterm Control PDFDocument13 pages1st Midterm Control PDFAlok KumarNo ratings yet
- The Cell CycleDocument72 pagesThe Cell CycleJerry Jeroum RegudoNo ratings yet
- Pearson Science TextbookDocument50 pagesPearson Science TextbookRick100% (2)
- Test - Embryology Practice Questions With Answers - QuizletDocument13 pagesTest - Embryology Practice Questions With Answers - QuizletAzizNo ratings yet
- Fundamentals of Bio Informatics Multiple Choice Question (GuruKpo)Document7 pagesFundamentals of Bio Informatics Multiple Choice Question (GuruKpo)GuruKPO100% (1)
- Arthroscopy of The Temporomandibular JointDocument44 pagesArthroscopy of The Temporomandibular JointDencyclopediaNo ratings yet
- Ottawa Charter For Health PromotionDocument5 pagesOttawa Charter For Health PromotionEleanorNo ratings yet
- In Vitro Gas Measuring Techniques Review HohenheimDocument21 pagesIn Vitro Gas Measuring Techniques Review HohenheimHelio LimaNo ratings yet
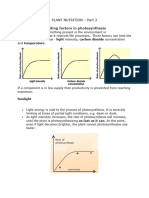
- PLANT NUTRITION Part 2Document10 pagesPLANT NUTRITION Part 2dineo kabasiaNo ratings yet
- Hill Stream AdaptationDocument5 pagesHill Stream AdaptationAdarsh SenNo ratings yet
- Human Blood GroupsDocument12 pagesHuman Blood GroupsAme Roxan AwidNo ratings yet
- Bioorganic & Medicinal Chemistry: Christoph Nitsche, Christian Steuer, Christian D. KleinDocument20 pagesBioorganic & Medicinal Chemistry: Christoph Nitsche, Christian Steuer, Christian D. KleinCiro LiraNo ratings yet
- Seed Production Technologyof Wheatand BarleyDocument33 pagesSeed Production Technologyof Wheatand BarleyAlemayehu TeshomeNo ratings yet
- Maculabatis Ambigua Sp. Nov., A New Whipray (Myliobatiformes: Dasyatidae) From The Western Indian OceanDocument14 pagesMaculabatis Ambigua Sp. Nov., A New Whipray (Myliobatiformes: Dasyatidae) From The Western Indian OceanIlham FajriNo ratings yet
- B05 QuestionsDocument5 pagesB05 QuestionsMauriya SenthilnathanNo ratings yet
- Catalog of Nearctic Chironomidae PDFDocument98 pagesCatalog of Nearctic Chironomidae PDFJumentoNo ratings yet
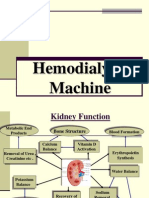
- Hemodialysis Machine12Document45 pagesHemodialysis Machine12Divya SoundarajanNo ratings yet
- Mudras For Awakening Chakras 19Document93 pagesMudras For Awakening Chakras 19NiravMakwanaNo ratings yet
- Study Guide Dna Rna No VirusDocument4 pagesStudy Guide Dna Rna No VirusLara AlbeeshyNo ratings yet
- Anxiety Lesson WorksheetDocument4 pagesAnxiety Lesson WorksheetAmisha VastaniNo ratings yet
- Megaloblastic Anaemia: Folic Acid and Vitamin B12 MetabolismDocument9 pagesMegaloblastic Anaemia: Folic Acid and Vitamin B12 MetabolismLuna100% (1)
- Psychology Core Concepts 7th Edition Zimbardo Test BankDocument78 pagesPsychology Core Concepts 7th Edition Zimbardo Test Banketiolatepuff33tj100% (31)
- Biology Paper 6 November October 2008 IGCSE Question Paper QPDocument12 pagesBiology Paper 6 November October 2008 IGCSE Question Paper QPStevo4975% (4)