Download as pdf or txt
You might also like
- Solutions To Haynie Ch. 2Document7 pagesSolutions To Haynie Ch. 2Dharin DetamaNo ratings yet
- Ap Unit 5Document25 pagesAp Unit 5Alecs JovellanosNo ratings yet
- Physics Assignment Document-Converted-Pages-DeletedDocument11 pagesPhysics Assignment Document-Converted-Pages-DeletedSUBERUS HEARTRISHANo ratings yet
- Chapter 8Document19 pagesChapter 8احمد غالبNo ratings yet
- Gibbs Paradox SolutionDocument11 pagesGibbs Paradox SolutioncraverNo ratings yet
- Chapter 20: Entropy and The Second Law of ThermodynamicsDocument61 pagesChapter 20: Entropy and The Second Law of ThermodynamicsMR éxypnosNo ratings yet
- 1 2 3 Literature Review 4 Experiment Objective 5 Methodology 6 Results 7 Discussions 8 Conclusion & Recommendations 9 References 10 AppendicesDocument16 pages1 2 3 Literature Review 4 Experiment Objective 5 Methodology 6 Results 7 Discussions 8 Conclusion & Recommendations 9 References 10 Appendicesmonkeystar12100% (3)
- Mole: 1 Mole of A Substance Contains Avogadro's Number (N 6.02E23)Document53 pagesMole: 1 Mole of A Substance Contains Avogadro's Number (N 6.02E23)Juan Carlos Gonzalez LNo ratings yet
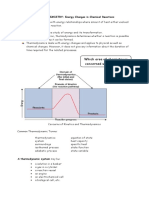
- Which Area of Chemistry Is Concerned With The Topic?: THERMOCHEMISTRY: Energy Changes in Chemical ReactionsDocument19 pagesWhich Area of Chemistry Is Concerned With The Topic?: THERMOCHEMISTRY: Energy Changes in Chemical ReactionsSahada KanapiyaNo ratings yet
- 11.0 Thermal PhysicsDocument42 pages11.0 Thermal PhysicsJoe MathewNo ratings yet
- Heat and TemperatureDocument9 pagesHeat and TemperatureAyush ChandekarNo ratings yet
- Introduction To Thermodynamics PDFDocument131 pagesIntroduction To Thermodynamics PDFtranhuutuongNo ratings yet
- Thermal Physics 12 GradeDocument42 pagesThermal Physics 12 GradeAmri Ramadhan D'classicNo ratings yet
- Research TopicDocument18 pagesResearch TopicmuhammadNo ratings yet
- Thermodynamic Unit 4Document10 pagesThermodynamic Unit 4Bhavani .SNo ratings yet
- Anneli Aitta 2006, "Iron Melting Curve With A Tricritical Point", Journal of Statistical MechanicsDocument17 pagesAnneli Aitta 2006, "Iron Melting Curve With A Tricritical Point", Journal of Statistical MechanicsNguyen Tri NguyenNo ratings yet
- Thermochemistry or Junior IntermideateDocument48 pagesThermochemistry or Junior IntermideateEvs GoudNo ratings yet
- Handout 9 ThermodynamicsDocument10 pagesHandout 9 ThermodynamicsMary Grace AcostaNo ratings yet
- Thermal PhysicsDocument49 pagesThermal PhysicsClaudia PinnaNo ratings yet
- Electrosdfghjkchemical Determination of GHSDocument7 pagesElectrosdfghjkchemical Determination of GHSluis carlos castilloNo ratings yet
- Thermochemistry: Purpose of The ExperimentDocument20 pagesThermochemistry: Purpose of The ExperimentAngel LacsonNo ratings yet
- 08 Thermochemistry 2bDocument19 pages08 Thermochemistry 2bchewazableNo ratings yet
- Solutions Manual Chapter17Document29 pagesSolutions Manual Chapter17더브레인코어과학관No ratings yet
- The Law of Entropy Invcrease - A Lab ExperimentDocument4 pagesThe Law of Entropy Invcrease - A Lab ExperimentINDUSTRIA BIOTECNOLÓGICA DEL SURNo ratings yet
- 312 Physics Eng Lesson10Document27 pages312 Physics Eng Lesson10AyushNo ratings yet
- CalorimetryDocument4 pagesCalorimetryRomin PhyRoyNo ratings yet
- AP Chemistry 2013-2014 Lab #13 - Hot Pack/Cold Pack Design ChallengeDocument4 pagesAP Chemistry 2013-2014 Lab #13 - Hot Pack/Cold Pack Design ChallengeAman GuptaNo ratings yet
- 154-156 (Chen)Document3 pages154-156 (Chen)alembrhanNo ratings yet
- Chap-11 Heat And Thermodynamics KeypointsDocument17 pagesChap-11 Heat And Thermodynamics Keypointsbb5299772No ratings yet
- Kumar Nussinov Biochem 2001 Mesophile Thermophile StabilityDocument14 pagesKumar Nussinov Biochem 2001 Mesophile Thermophile StabilityJames NanorNo ratings yet
- Chem 16Document8 pagesChem 16Adi SoNo ratings yet
- Latent Heat of VaporizationDocument11 pagesLatent Heat of VaporizationEsther Faith GabrielNo ratings yet
- Module 3 - EntropyDocument80 pagesModule 3 - EntropyDhruvanth SJNo ratings yet
- Assignment Class: Xi (Chemistry) ThermodynamicsDocument3 pagesAssignment Class: Xi (Chemistry) Thermodynamicskeshavjain7No ratings yet
- Chapter - 6 ThermodynamicDocument7 pagesChapter - 6 ThermodynamicManan TyagiNo ratings yet
- Thermodynamic Processes: (3) Isobaric Processes: Those Processes Which Take Place at Constant Pressure Are CalledDocument16 pagesThermodynamic Processes: (3) Isobaric Processes: Those Processes Which Take Place at Constant Pressure Are CalledKarwan AliNo ratings yet
- TECreview ImprovecoefficientDocument17 pagesTECreview ImprovecoefficienthalimberdiNo ratings yet
- LP2Document23 pagesLP2Vinz TaquiquiNo ratings yet
- 11 12 Thermal-PhysicsDocument2 pages11 12 Thermal-PhysicshokaiyiNo ratings yet
- Er 991Document16 pagesEr 991Grill meisterNo ratings yet
- Heat of Reaction Sum 2011Document11 pagesHeat of Reaction Sum 2011Seohyun ChoNo ratings yet
- Thermodynamics: 222 PHYSDocument24 pagesThermodynamics: 222 PHYSAdhanom G.100% (1)
- Thermodynamics: Lecture 2 Review of Lecture 1Document10 pagesThermodynamics: Lecture 2 Review of Lecture 1Lexter Gomez GabicaNo ratings yet
- Boiling & CondensationDocument22 pagesBoiling & CondensationNITISH KUMARNo ratings yet
- EntropyDocument21 pagesEntropycusgakungaNo ratings yet
- 50 Lab Calorimetry of A Cold Pack GolestanehDocument10 pages50 Lab Calorimetry of A Cold Pack GolestanehnanaNo ratings yet
- SECOND LAW of ThermodynamicsDocument23 pagesSECOND LAW of ThermodynamicsDianne VillanuevaNo ratings yet
- Kennard E.H. - Entropy, Reversible Processes and Thermo-Couples (1932)Document5 pagesKennard E.H. - Entropy, Reversible Processes and Thermo-Couples (1932)Juan Sebastian SánchezNo ratings yet
- Cap 5 Examen (01-13)Document14 pagesCap 5 Examen (01-13)RuthNo ratings yet
- Heat and ThermodynamicDocument10 pagesHeat and ThermodynamicAmina UroojNo ratings yet
- Chapter20 1a MHDocument61 pagesChapter20 1a MHElla ZambraNo ratings yet
- Experiment 6 Formal ReportDocument9 pagesExperiment 6 Formal ReportmegmayorNo ratings yet
- Physics 2 1Document104 pagesPhysics 2 1Kimberly GonzalesNo ratings yet
- MME 2003 Metallurgical Thermodynamic: The Second Law of Thermodynamics and EntropyDocument40 pagesMME 2003 Metallurgical Thermodynamic: The Second Law of Thermodynamics and Entropycandost altınbaşNo ratings yet
- 2 Laws of ThermodynamicsDocument86 pages2 Laws of ThermodynamicsEDENI100% (1)
- Part 1. - Chem - ThermodynamicsDocument64 pagesPart 1. - Chem - ThermodynamicsTaymeng LyNo ratings yet
- Practice Makes Perfect in Chemistry: The Physical Behavior of MatterFrom EverandPractice Makes Perfect in Chemistry: The Physical Behavior of MatterRating: 5 out of 5 stars5/5 (1)
- “Foundations to Flight: Mastering Physics from Curiosity to Confidence: Cipher 4”: “Foundations to Flight: Mastering Physics from Curiosity to Confidence, #4From Everand“Foundations to Flight: Mastering Physics from Curiosity to Confidence: Cipher 4”: “Foundations to Flight: Mastering Physics from Curiosity to Confidence, #4Rating: 5 out of 5 stars5/5 (1)
- SSC Thermal EngineeringDocument47 pagesSSC Thermal EngineeringSteph Dela MujerNo ratings yet
- ABB Variable Frequency DriveDocument5 pagesABB Variable Frequency DriveAdam MuhammadNo ratings yet
- HBL NCPP LeafletDocument2 pagesHBL NCPP LeafletSunil KumawatNo ratings yet
- 0206 MACH7-Regelung Engl PDFDocument1 page0206 MACH7-Regelung Engl PDFSyed Mohammed Hussain100% (1)
- 2023-2024-Class XI-Physics-Part 5-AWDocument10 pages2023-2024-Class XI-Physics-Part 5-AWjimmyemandeeNo ratings yet
- RdsoDocument7 pagesRdsoMrinmy ChakrabortyNo ratings yet
- W-250P-60 SERIES: Polycrystalline Silicon Solar ModulesDocument1 pageW-250P-60 SERIES: Polycrystalline Silicon Solar ModulesLamagia OrtizNo ratings yet
- 7 Ecotel Outdoor v4Document4 pages7 Ecotel Outdoor v4Cesar PrietoNo ratings yet
- Lecture - 01 - PPT PS IDocument67 pagesLecture - 01 - PPT PS ITsega Solomon Kidane100% (7)
- MELCsDocument27 pagesMELCsRowena Sta MariaNo ratings yet
- HAGER AutomationProductsDocument32 pagesHAGER AutomationProductsMinos AsimakisNo ratings yet
- 230v LED Driver Circuit Diagram, Working and ApplicationsDocument8 pages230v LED Driver Circuit Diagram, Working and ApplicationsAbbas Sabbar GhaliNo ratings yet
- Theory of Operation of Single Phase Transformer - Custom Coils - CLIENTDocument8 pagesTheory of Operation of Single Phase Transformer - Custom Coils - CLIENTcustomcoilsNo ratings yet
- Analysis of The C Swan Bands As A Thermometric Probe in CO Microwave PlasmasDocument26 pagesAnalysis of The C Swan Bands As A Thermometric Probe in CO Microwave PlasmasematlisNo ratings yet
- Switchgear and Switchboard Inspection and Testing GuideDocument22 pagesSwitchgear and Switchboard Inspection and Testing GuideArif KhanNo ratings yet
- Blackout in PakistanDocument6 pagesBlackout in Pakistansyed Mujtaba hassanNo ratings yet
- NYS Clean Energy Standard NoticeDocument2 pagesNYS Clean Energy Standard NoticeRochester Democrat and ChronicleNo ratings yet
- Hence To Keep The CT Burden Lower, CT With 1A Secondary Is PreferableDocument1 pageHence To Keep The CT Burden Lower, CT With 1A Secondary Is PreferableAjay PatilNo ratings yet
- MPL15SXDocument5 pagesMPL15SXDragos ComanNo ratings yet
- A Dynamic Fleet Model of U.S Light-Duty Vehicle Lightweighting and Associated Greenhouse Gas Emissions From 2016 To 2050Document10 pagesA Dynamic Fleet Model of U.S Light-Duty Vehicle Lightweighting and Associated Greenhouse Gas Emissions From 2016 To 2050George FernandezNo ratings yet
- Nuclear and Space Radiation Effects On Materials: Nasa Space Vehicle Design CriteriaDocument48 pagesNuclear and Space Radiation Effects On Materials: Nasa Space Vehicle Design CriteriapitcromNo ratings yet
- Effectiveness Matrix: Bore and Stroke of 50mm X 49.5mmDocument6 pagesEffectiveness Matrix: Bore and Stroke of 50mm X 49.5mmAakash HoskotiNo ratings yet
- Final Prds RRRRDocument12 pagesFinal Prds RRRRBanjara JabardasthNo ratings yet
- Indoor RMU ManualDocument20 pagesIndoor RMU Manualhardeepsingh_08No ratings yet
- Undra Undra Undra Undra: ChileDocument1 pageUndra Undra Undra Undra: ChileValentin Bustos MolinaNo ratings yet
- PV SF2 LayoutDocument3 pagesPV SF2 LayoutrrrrrNo ratings yet
- Fuji TK26 016TN 22ADocument13 pagesFuji TK26 016TN 22AtokusanNo ratings yet
- 07 Opportunities For PoA in Energy Efficiency by Konrad Von RitterDocument11 pages07 Opportunities For PoA in Energy Efficiency by Konrad Von RitterThe Outer MarkerNo ratings yet
- Conclusion of Nuclear Accident at Fukushima DaiichiDocument2 pagesConclusion of Nuclear Accident at Fukushima DaiichiEnformableNo ratings yet
- Volume 2-Pkg 3 385 Tender Sub StationDocument446 pagesVolume 2-Pkg 3 385 Tender Sub StationKrishna Manandhar100% (1)