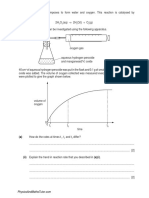
Download as pdf or txt
You might also like
- Student Exploration: TitrationDocument7 pagesStudent Exploration: TitrationMF - 11AK 827776 Central Peel SSNo ratings yet
- Poly EneDocument3 pagesPoly EneMohammed TarekNo ratings yet
- Pi Facial Selectivity in Diels Alder ReactionsDocument12 pagesPi Facial Selectivity in Diels Alder ReactionsSatyaki MajumdarNo ratings yet
- Jacs AsapDocument2 pagesJacs AsapMohamadMostafaviNo ratings yet
- 2005 Electrophilic BehaviorDocument23 pages2005 Electrophilic BehaviorlisusedNo ratings yet
- Tmp747e TMPDocument5 pagesTmp747e TMPFrontiersNo ratings yet
- Teresita Maldonando, Eduardo Martínez-González, Carlos FrontanaDocument1 pageTeresita Maldonando, Eduardo Martínez-González, Carlos FrontanaartedlcNo ratings yet
- 13 1Document4 pages13 1Йоханн БуренковNo ratings yet
- Spino1998 Journal of Organic Chemistry, 1998, Vol. 63, # 15, P. 5283 - 5287Document5 pagesSpino1998 Journal of Organic Chemistry, 1998, Vol. 63, # 15, P. 5283 - 5287SpazzaturaNo ratings yet
- Conformational Energy From The Oxidation Kinetics of Poly (3,4-Ethylenedioxythiophene) FilmsDocument8 pagesConformational Energy From The Oxidation Kinetics of Poly (3,4-Ethylenedioxythiophene) FilmsaneicouboiNo ratings yet
- Oxime DecompositionDocument6 pagesOxime DecompositionAmanda CamillaNo ratings yet
- Gold (I) - Catalyzed Intermolecular (2+2) Cycloaddition of Alkynes With AlkenesDocument3 pagesGold (I) - Catalyzed Intermolecular (2+2) Cycloaddition of Alkynes With Alkenesnestor pelaez restrepoNo ratings yet
- tmpB9C3 TMPDocument11 pagestmpB9C3 TMPFrontiersNo ratings yet
- Lanthanum Tricyanide-Catalyzed Acyl Silane - Ketone Benzoin Additions and Kinetic Resolution of Resultant R-SilyloxyketonesDocument9 pagesLanthanum Tricyanide-Catalyzed Acyl Silane - Ketone Benzoin Additions and Kinetic Resolution of Resultant R-SilyloxyketonesDiogo DiasNo ratings yet
- Nihms 1683173Document10 pagesNihms 1683173Tetty Arsety GuluhNo ratings yet
- 1 s2.0 S1093326323000165 MainDocument13 pages1 s2.0 S1093326323000165 MainDanso MichaelNo ratings yet
- Organic SynthesisDocument35 pagesOrganic SynthesisAshok PareekNo ratings yet
- Modular Terpene Synthesis Enabled by Mild Electrochemical CouplingsDocument18 pagesModular Terpene Synthesis Enabled by Mild Electrochemical Couplingsgabriel martinezNo ratings yet
- Theoretical Investigation of Regiodivergent AdditiDocument16 pagesTheoretical Investigation of Regiodivergent AdditiLavinia KassabNo ratings yet
- Stereochemical Aspects of Organic SynthesisDocument9 pagesStereochemical Aspects of Organic SynthesisNoor NNo ratings yet
- Theoretical Study On The Alkylation of O-Xylene WithDocument23 pagesTheoretical Study On The Alkylation of O-Xylene WithPrasad BolisettyNo ratings yet
- 8a Miller1995Document2 pages8a Miller1995Tu NaruNo ratings yet
- Pericyclic ReactionsDocument5 pagesPericyclic ReactionsNurul HidayahNo ratings yet
- Full Paper: Synthesis and EPR Studies of 2'-Deoxyuridines With Alkynyl, Rodlike LinkagesDocument9 pagesFull Paper: Synthesis and EPR Studies of 2'-Deoxyuridines With Alkynyl, Rodlike Linkagessrinivasarao meneniNo ratings yet
- PD Isomerization Mazet JACS 2016Document7 pagesPD Isomerization Mazet JACS 2016Thierry GranierNo ratings yet
- Stereoelectronic Effects: A Bridge Between Structure and ReactivityFrom EverandStereoelectronic Effects: A Bridge Between Structure and ReactivityNo ratings yet
- Electrochemical Synthesis of Azo Azulene FilmsDocument6 pagesElectrochemical Synthesis of Azo Azulene FilmsAdina NițăNo ratings yet
- 10 1002@ (Sici) 1521-3765 (19990401) 5:4 1320::aid-Chem1320 3 0 Co 2-#Document11 pages10 1002@ (Sici) 1521-3765 (19990401) 5:4 1320::aid-Chem1320 3 0 Co 2-#Yassine SabekNo ratings yet
- Electropol Edot Sds TritonDocument8 pagesElectropol Edot Sds TritonGonzalo FenoyNo ratings yet
- C 1 JM 10649 JDocument10 pagesC 1 JM 10649 JHamada BouchinaNo ratings yet
- Informe - Titulacion Potenciometrica Del Acido FosforicoDocument10 pagesInforme - Titulacion Potenciometrica Del Acido FosforicoScarlet Jacqueline Salas CalvoNo ratings yet
- Organometallic Chemistry at The Nanoscale. Dendrimers For Redox Processes and CatalysisDocument21 pagesOrganometallic Chemistry at The Nanoscale. Dendrimers For Redox Processes and CatalysisVeselina GeorgievaNo ratings yet
- Manlio T20056443Document9 pagesManlio T20056443api-19793040No ratings yet
- Regioselective Substitution at The 1 3 and 6 8 Positions of Pyrene For The Construction of Small Dipolar MoleculesDocument6 pagesRegioselective Substitution at The 1 3 and 6 8 Positions of Pyrene For The Construction of Small Dipolar MoleculesRaviNo ratings yet
- Diels Alder ReactionDocument10 pagesDiels Alder ReactionagrimmittalNo ratings yet
- Notes Lecture 1 Conformational AnalysisDocument18 pagesNotes Lecture 1 Conformational AnalysisDianing Wismarani Putri100% (1)
- Schalenbach 2016 J. Electrochem. Soc. 163 F3197Document13 pagesSchalenbach 2016 J. Electrochem. Soc. 163 F3197Avishek JaiswalNo ratings yet
- Diels-Alder Reaction: MechanismDocument5 pagesDiels-Alder Reaction: MechanismJavier RamirezNo ratings yet
- Well-De Fined Nanographene Rhenium Complex As An E Cient Electrocatalyst and Photocatalyst For Selective CO ReductionDocument4 pagesWell-De Fined Nanographene Rhenium Complex As An E Cient Electrocatalyst and Photocatalyst For Selective CO ReductionZUNENo ratings yet
- Electropolymerization of Pyrrole and Electrochemical Study of Polypyrrole: 1. Evidence For Structural Diversity of PolypyrroleDocument16 pagesElectropolymerization of Pyrrole and Electrochemical Study of Polypyrrole: 1. Evidence For Structural Diversity of PolypyrroleEmmanuel Avalos HuarteNo ratings yet
- Kin 20149Document6 pagesKin 20149Bruna ButkeNo ratings yet
- Materials 03 04773Document11 pagesMaterials 03 04773Taufik HidayatNo ratings yet
- C16Document12 pagesC16lacewingNo ratings yet
- Heterogeneous Asymmetric Diels-Alder Reactions Using A Copper-Chiral Bis (Oxazoline) Complex Immobilized On Mesoporous SilicaDocument5 pagesHeterogeneous Asymmetric Diels-Alder Reactions Using A Copper-Chiral Bis (Oxazoline) Complex Immobilized On Mesoporous SilicaJC Jane BarnesNo ratings yet
- Methods To Study The Ionic Conductivity of Polymeric Electrolytes Using A.C. Impedance SpectrosDocument8 pagesMethods To Study The Ionic Conductivity of Polymeric Electrolytes Using A.C. Impedance Spectrosseung leeNo ratings yet
- Direct Iminization of PEEK: Ioannis Manolakis, Paul Cross, and Howard M. ColquhounDocument4 pagesDirect Iminization of PEEK: Ioannis Manolakis, Paul Cross, and Howard M. ColquhounPedro RosaNo ratings yet
- Nucleophilic Aromatic Substitution Reactions Described by The Local Electron Attachment EnergyDocument37 pagesNucleophilic Aromatic Substitution Reactions Described by The Local Electron Attachment EnergyYusuf Bayu AjiNo ratings yet
- FerrocyanideDocument9 pagesFerrocyanideAlberto AyónNo ratings yet
- Stereo SelectivityDocument9 pagesStereo SelectivityrashidNo ratings yet
- Preparation of 2-Alkylidene Oxetanes: An Investigation of The Paterno-Biichi Reaction Between Aliphatic Aldehydes and AllenesDocument4 pagesPreparation of 2-Alkylidene Oxetanes: An Investigation of The Paterno-Biichi Reaction Between Aliphatic Aldehydes and AllenesSaurav PaulNo ratings yet
- High T, Ambipolar, and Near-Infrared Electrochromic Anthraquinone-Based Aramids With Intervalence Charge-Transfer BehaviorDocument9 pagesHigh T, Ambipolar, and Near-Infrared Electrochromic Anthraquinone-Based Aramids With Intervalence Charge-Transfer BehaviorManthan JainNo ratings yet
- 10 1002@cssc 202000098Document8 pages10 1002@cssc 202000098juliana perez ordoñezNo ratings yet
- 11 Redox Comp-DispDocument7 pages11 Redox Comp-DisppathisharmaNo ratings yet
- Tetrahedron 2010 66 10 01902 - 01910 CiclicoDocument9 pagesTetrahedron 2010 66 10 01902 - 01910 Ciclicoteodoro11No ratings yet
- ad65cdd43c3dff81d7451e4237efd5ebDocument2 pagesad65cdd43c3dff81d7451e4237efd5ebguiburNo ratings yet
- tmpC462 TMPDocument7 pagestmpC462 TMPFrontiersNo ratings yet
- Paterno Buchi ReactionDocument35 pagesPaterno Buchi ReactionHarish Chopra100% (1)
- Zhou 1992Document8 pagesZhou 1992luuphuongNo ratings yet
- Liu2009 PDFDocument6 pagesLiu2009 PDFpolypolyyNo ratings yet
- Electrically Conducting Three-Dimensional (Octacyano Phthalocyaninato Polysiloxane) PolymerDocument6 pagesElectrically Conducting Three-Dimensional (Octacyano Phthalocyaninato Polysiloxane) PolymerZachary AronsNo ratings yet
- Computational Methods in Lanthanide and Actinide ChemistryFrom EverandComputational Methods in Lanthanide and Actinide ChemistryMichael DolgNo ratings yet
- The Cellular Transport Week 6Document28 pagesThe Cellular Transport Week 6Angel Mae Masa FloresNo ratings yet
- Weathering of Rocks - LectureDocument21 pagesWeathering of Rocks - LectureSharyl Plan SarominesNo ratings yet
- Organic Chemistry: Course Code: SKO 3033 Semester 2 Sessions 2020/2021 Id Number and NameDocument6 pagesOrganic Chemistry: Course Code: SKO 3033 Semester 2 Sessions 2020/2021 Id Number and NameSITI HUMAIRAH BINTI HAMZAHNo ratings yet
- Ncert Page Wise Q Structural OrganizationDocument16 pagesNcert Page Wise Q Structural OrganizationSagarNo ratings yet
- JW GC and HPLC (Notes On Othe Chromatographic Techinques Are Absent)Document21 pagesJW GC and HPLC (Notes On Othe Chromatographic Techinques Are Absent)Greatness AgwazeNo ratings yet
- 9.1 Multiple-Choice and Bimodal QuestionsDocument18 pages9.1 Multiple-Choice and Bimodal QuestionsQuốc Thắng NguyễnNo ratings yet
- March 2016 (v2) QP - Paper 3 CIE Chemistry IGCSE - WatermarkDocument16 pagesMarch 2016 (v2) QP - Paper 3 CIE Chemistry IGCSE - WatermarkAla'No ratings yet
- Importance of Chemistry in NutritionDocument1 pageImportance of Chemistry in Nutritionchiles_6350% (2)
- Past Paper Bmat 2021 Section 2Document24 pagesPast Paper Bmat 2021 Section 2Kaew ThanidaNo ratings yet
- Glaxo Vol IDocument164 pagesGlaxo Vol IPrakash WarrierNo ratings yet
- Fixation and Fixatives in HistopathologyDocument13 pagesFixation and Fixatives in HistopathologypayalNo ratings yet
- Grade 9 2nd Quarter Module 4 Carbon A Special Element FinalizedDocument26 pagesGrade 9 2nd Quarter Module 4 Carbon A Special Element FinalizedAkisha Jen CalicdanNo ratings yet
- RM 1 Mix QUESTION 1 230418 201559Document7 pagesRM 1 Mix QUESTION 1 230418 201559Jash ShahNo ratings yet
- June 2022 (9701 - 12) QPDocument20 pagesJune 2022 (9701 - 12) QPHung Mang ThiNo ratings yet
- Class 12chemistry - Alcohol, Phenol and Ether - McqsDocument22 pagesClass 12chemistry - Alcohol, Phenol and Ether - McqsShypackofcheetosNo ratings yet
- Industrial Crops & Products: A A B C D e F GDocument13 pagesIndustrial Crops & Products: A A B C D e F GDerek ZoolanderNo ratings yet
- 1 Hydrogen Peroxide Decomposes To Form Water and Oxygen. This Reaction Is Catalysed byDocument9 pages1 Hydrogen Peroxide Decomposes To Form Water and Oxygen. This Reaction Is Catalysed byKelvin DenhereNo ratings yet
- Light-Dependent Herbicides - An OverviewDocument12 pagesLight-Dependent Herbicides - An OverviewCatherine TangNo ratings yet
- Ishihara2019 MPCDocument11 pagesIshihara2019 MPCKrisna MertaNo ratings yet
- CBSE Class 10 Science - Acids Bases and SaltsDocument2 pagesCBSE Class 10 Science - Acids Bases and SaltsAnuska SaxenaNo ratings yet
- Class - XII (Frequntly Asked Topics)Document13 pagesClass - XII (Frequntly Asked Topics)yrh6qgd2xnNo ratings yet
- HW - Carbohydrate Metabolism II & Lipid MetabolismDocument2 pagesHW - Carbohydrate Metabolism II & Lipid MetabolismyanNo ratings yet
- Sono-Chemical Leaching of UraniumDocument8 pagesSono-Chemical Leaching of UraniumLucas EduardoNo ratings yet
- Cenabre, Sedeno, Ybanez Quantitative ResearchDocument29 pagesCenabre, Sedeno, Ybanez Quantitative ResearchMarco cenabreNo ratings yet
- Dowex™ Monosphere™ 88: Product Type Matrix Functional GroupDocument2 pagesDowex™ Monosphere™ 88: Product Type Matrix Functional GroupBrianNo ratings yet
- 8 AshLand Hair Care ListDocument32 pages8 AshLand Hair Care Listcontentdrive4 drive4No ratings yet
- Lecture Planner - Organic Chemistry - Prayas FastrackDocument2 pagesLecture Planner - Organic Chemistry - Prayas FastrackNIKHIL PATNAIKNo ratings yet
- Test For Carboxylic Acids and Derivatives - Online ClassDocument3 pagesTest For Carboxylic Acids and Derivatives - Online Classjoseph cyron solidumNo ratings yet
- Assay Principle For Different Inorg CompsDocument2 pagesAssay Principle For Different Inorg Compsdinesh_thakkar_5No ratings yet