Download as pdf or txt
You might also like
- Metallic Oxides by GoodenoughDocument255 pagesMetallic Oxides by Goodenoughmuk_hawkNo ratings yet
- PlasmonssDocument44 pagesPlasmonssKim Trinh Tran ThiNo ratings yet
- Grade 12 Lesson 10Document60 pagesGrade 12 Lesson 10Van CometaNo ratings yet
- General Static Polarizability in Spherical Neutral Metal Clusters and Fullerenes Within Thomas-Fermi TheoryDocument17 pagesGeneral Static Polarizability in Spherical Neutral Metal Clusters and Fullerenes Within Thomas-Fermi TheoryThomas PaladeNo ratings yet
- Crystal Field Theory (CFT)Document15 pagesCrystal Field Theory (CFT)veronicaNo ratings yet
- Electronic Spectra of ComplexesDocument82 pagesElectronic Spectra of Complexesirembasar2000No ratings yet
- CFTDocument15 pagesCFTGaurav BothraNo ratings yet
- Electronic Band Structure and Lattice Distortion in Perovskite Transition-Metal OxidesDocument3 pagesElectronic Band Structure and Lattice Distortion in Perovskite Transition-Metal Oxidesanon_452078312No ratings yet
- 1 s2.0 S0927796X02001043 MainDocument56 pages1 s2.0 S0927796X02001043 MainpescaofritoNo ratings yet
- Crystal Field TheoryDocument7 pagesCrystal Field TheoryD GNo ratings yet
- Class 4Document13 pagesClass 4Muskan BiswalNo ratings yet
- Overview of Crystal Field Theory AnalysisDocument11 pagesOverview of Crystal Field Theory Analysiskashif nadeem SaabriNo ratings yet
- CFT PDFDocument20 pagesCFT PDFRUFAS KANIKANTINo ratings yet
- Hard Soft Acid Base Theory or HSABDocument9 pagesHard Soft Acid Base Theory or HSABAMAN JATNo ratings yet
- Spectra of TransitionDocument43 pagesSpectra of TransitionAditya MahakalNo ratings yet
- Electronic Spectroscopy of Transition Metal CompoundsDocument24 pagesElectronic Spectroscopy of Transition Metal CompoundsBruno Ramos de LimaNo ratings yet
- Edward L Hamilton, Chris H Greene and H R Sadeghpour - Shape-Resonance-Induced Long-Range Molecular Rydberg StatesDocument8 pagesEdward L Hamilton, Chris H Greene and H R Sadeghpour - Shape-Resonance-Induced Long-Range Molecular Rydberg StatesItama23No ratings yet
- Density Functional Theory and Experimental Study of The Electronic Structure and Transport Properties of La, V, NB, and Ta Doped Srtio3Document12 pagesDensity Functional Theory and Experimental Study of The Electronic Structure and Transport Properties of La, V, NB, and Ta Doped Srtio3Shridhar MathadNo ratings yet
- Ligand Field N MOTDocument12 pagesLigand Field N MOTLata Sharma100% (1)
- Crystal Field Theory - HandoutDocument7 pagesCrystal Field Theory - HandoutHastings FrazerNo ratings yet
- CH2422 Electronic Spectra of Transition MetalsDocument6 pagesCH2422 Electronic Spectra of Transition MetalsJohnNo ratings yet
- Oxygen Vacancies Effect On The Magnetic Properties of SrTiFeO3Document23 pagesOxygen Vacancies Effect On The Magnetic Properties of SrTiFeO3solisiusNo ratings yet
- Crystal Field TheoryDocument6 pagesCrystal Field TheoryRamashish ChoudharyNo ratings yet
- Crystal Field Theory - NURDocument5 pagesCrystal Field Theory - NURNurhajrahNo ratings yet
- Crystal Field TheoryDocument5 pagesCrystal Field TheoryDebmalya Gharai100% (1)
- tmp6ECC TMPDocument3 pagestmp6ECC TMPFrontiersNo ratings yet
- Electron-Electron and Electron-Phonon Interactions in Graphene On A Semiconductor Substrate: Simple EstimationsDocument6 pagesElectron-Electron and Electron-Phonon Interactions in Graphene On A Semiconductor Substrate: Simple EstimationsNicolás Parra AvilaNo ratings yet
- Unit 5 Coordination Chemistry: Graduate Center Inorganic Chemistry (Fall 2012)Document39 pagesUnit 5 Coordination Chemistry: Graduate Center Inorganic Chemistry (Fall 2012)Carlos Cesar Lopez SuarezNo ratings yet
- BookDocument44 pagesBookDr-Mandeep SinghNo ratings yet
- 2 TM Class PPT - BB Aug-Nov 2019Document28 pages2 TM Class PPT - BB Aug-Nov 2019StephenNo ratings yet
- Magnetochemistry 08 00144Document30 pagesMagnetochemistry 08 00144solisiusNo ratings yet
- Long-Range Proximity Effects in Superconductor-Ferromagnet StructuresDocument4 pagesLong-Range Proximity Effects in Superconductor-Ferromagnet StructurespanzhanghiggsNo ratings yet
- Crystal Field TheoryDocument9 pagesCrystal Field TheoryMa'arif A. SyafiiNo ratings yet
- Eldas AnorDocument8 pagesEldas AnorHaryadi Nugraha PutraNo ratings yet
- Electronic SpectraDocument22 pagesElectronic SpectraVishnu ChariNo ratings yet
- Atomic B1 ProblemsDocument8 pagesAtomic B1 ProblemsSimon Maxwell-StewartNo ratings yet
- 1 s2.0 S0277538721003818 MainDocument11 pages1 s2.0 S0277538721003818 MainMoromi NathNo ratings yet
- Electronic Spectra of TM ComplexesDocument35 pagesElectronic Spectra of TM Complexesbits_who_am_iNo ratings yet
- Ligand Effects in Heterogeneous Catalysis and ElectrochemistryDocument5 pagesLigand Effects in Heterogeneous Catalysis and ElectrochemistryLuca BrunoNo ratings yet
- Chapter 19 D-Metal Complexes: Electronic Structure and SpectraDocument70 pagesChapter 19 D-Metal Complexes: Electronic Structure and SpectraSadaf KhanNo ratings yet
- EsrDocument5 pagesEsrVirendra Singh Rajput100% (1)
- Applsci 10 05730Document13 pagesApplsci 10 05730fit threesNo ratings yet
- Surface Symmetry-Breaking and Strain Effects On Orbital Occupancy in Transition Metal Perovskite Epitaxial FilmsDocument7 pagesSurface Symmetry-Breaking and Strain Effects On Orbital Occupancy in Transition Metal Perovskite Epitaxial FilmsAshok KumarNo ratings yet
- Journal For Mathematical StudiesDocument18 pagesJournal For Mathematical StudiesRahul AmbawataNo ratings yet
- Electronic Spectra of Transition MetalDocument39 pagesElectronic Spectra of Transition MetaljohnwilliamsNo ratings yet
- VBTDocument40 pagesVBTLohith LoliNo ratings yet
- Plasmonic Physics of 2D Crystalline MaterialsDocument30 pagesPlasmonic Physics of 2D Crystalline Materials유재원No ratings yet
- PhysRevB.100.121113 AcceptedDocument7 pagesPhysRevB.100.121113 AcceptedDipesh GuptaNo ratings yet
- Bonding Patterns in Transition Metal Clusters: A. CeulemansDocument8 pagesBonding Patterns in Transition Metal Clusters: A. CeulemansSaurav PaulNo ratings yet
- Surface Plasmon - Dielectric Function Versus WavelengthDocument44 pagesSurface Plasmon - Dielectric Function Versus WavelengthEmerson KohlrauschNo ratings yet
- Storey Sochi 2012Document13 pagesStorey Sochi 2012taha_sochiNo ratings yet
- Ligand Field StrengthDocument24 pagesLigand Field StrengthIrvandar NurviandyNo ratings yet
- Seminar and Technical Writing 5th semestar-521CY1019Document8 pagesSeminar and Technical Writing 5th semestar-521CY1019RamakrishnaNo ratings yet
- Comparison of Octahedral and Tetrahedral FieldsDocument30 pagesComparison of Octahedral and Tetrahedral FieldsShubham Kumar100% (1)
- L'Kansition Metals Alloys: &F-PKJDocument1 pageL'Kansition Metals Alloys: &F-PKJKetanNo ratings yet
- Amorphous Semiconductors: Structural, Optical, and Electronic PropertiesFrom EverandAmorphous Semiconductors: Structural, Optical, and Electronic PropertiesNo ratings yet
- Electron Beam-Specimen Interactions and Simulation Methods in MicroscopyFrom EverandElectron Beam-Specimen Interactions and Simulation Methods in MicroscopyNo ratings yet
- Syllabus ICSE Chemistry-10Document4 pagesSyllabus ICSE Chemistry-10SJNo ratings yet
- Odisha Chemistry 11 Syllabus For IscDocument5 pagesOdisha Chemistry 11 Syllabus For IscSJNo ratings yet
- CVR Conceptual Chemistry 11 Vol 1Document1 pageCVR Conceptual Chemistry 11 Vol 1SJNo ratings yet
- CVR CBSE-Physics 9thDocument1 pageCVR CBSE-Physics 9thSJNo ratings yet
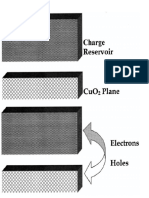
- Electronic Layers in Copper Oxide SuperconductorsDocument1 pageElectronic Layers in Copper Oxide SuperconductorsSJNo ratings yet
- CVR CBSE - Physics10Document1 pageCVR CBSE - Physics10SJNo ratings yet
- CVR CBSE Chemistry Class-9Document1 pageCVR CBSE Chemistry Class-9SJNo ratings yet
- Life Cycle Framework For Clean Manufacturing TechnologyDocument1 pageLife Cycle Framework For Clean Manufacturing TechnologySJNo ratings yet
- Chemists Control The HurricaneDocument1 pageChemists Control The HurricaneSJNo ratings yet
- Physical Processes of Luminescence On Isolated CentresDocument1 pagePhysical Processes of Luminescence On Isolated CentresSJNo ratings yet
- Clock PuzzlesDocument5 pagesClock PuzzlesSJ100% (1)
- Arithmetic & Algebraic Problems - pt-1Document4 pagesArithmetic & Algebraic Problems - pt-1SJ100% (1)
- Speed and Distance PuzzlesDocument7 pagesSpeed and Distance PuzzlesSJNo ratings yet
- Irish JaunttDocument1 pageIrish JaunttSJNo ratings yet
- Age PuzzlesDocument1 pageAge PuzzlesSJNo ratings yet
- IntroDocument1 pageIntroSJNo ratings yet
- Record CountDocument1 pageRecord CountSJNo ratings yet
- ScopeDocument1 pageScopeSJNo ratings yet
- NanostrDocument1 pageNanostrSJNo ratings yet
- Stoichemtry Unit Test Answer KeyDocument11 pagesStoichemtry Unit Test Answer KeyAnred CabahugNo ratings yet
- LNDHN Stondatd Specification For Rubber Conveyor and Elevator BeltingDocument5 pagesLNDHN Stondatd Specification For Rubber Conveyor and Elevator BeltingToufik KarimNo ratings yet
- E1 Methanol Handbook v2013Document40 pagesE1 Methanol Handbook v2013kenoly123100% (1)
- Grade of Concrete - Concrete Classification - Types of Concrete Mix - M25 Ratio - M30 Concrete Mix Ratio - M20 RatioDocument2 pagesGrade of Concrete - Concrete Classification - Types of Concrete Mix - M25 Ratio - M30 Concrete Mix Ratio - M20 RatioJayvee DignosNo ratings yet
- NPL-NG-P140001-BM-752 Pipe Material Class Summary - Rev 2.0Document38 pagesNPL-NG-P140001-BM-752 Pipe Material Class Summary - Rev 2.0Kehinde AdebayoNo ratings yet
- Farmhouse Dog Bowl Stand: RB NADocument5 pagesFarmhouse Dog Bowl Stand: RB NAO'Neil JonesNo ratings yet
- TSuprem4 ManualDocument920 pagesTSuprem4 ManualshatilaNo ratings yet
- 1 A Comprehensive Review of Different Types of Solar Photovoltaic Cells and Their ApplicationsDocument19 pages1 A Comprehensive Review of Different Types of Solar Photovoltaic Cells and Their ApplicationsHaider AddewanyNo ratings yet
- Birring NDE Center FlyerDocument1 pageBirring NDE Center FlyerMg MgNo ratings yet
- Monoproof Pu 1000: Single Component, Aliphatic, Water Based Exterior Pu Modified, Flexible Water Proofing MembraneDocument5 pagesMonoproof Pu 1000: Single Component, Aliphatic, Water Based Exterior Pu Modified, Flexible Water Proofing MembraneMonarch DigitalNo ratings yet
- MSDS Creatinine JaffeDocument4 pagesMSDS Creatinine JaffePrinceRCCastroNo ratings yet
- Bezinal3000 2010Document2 pagesBezinal3000 2010tanto_deep_15No ratings yet
- Acc. Valves in - To PED N Air Sepa On The Ba Aration Un Asis of AD Nits D 2000Document4 pagesAcc. Valves in - To PED N Air Sepa On The Ba Aration Un Asis of AD Nits D 2000sriramNo ratings yet
- VITABLOCS Working Instructions 1769EDocument56 pagesVITABLOCS Working Instructions 1769EAlexander L. Contreras PairaNo ratings yet
- Fema 450 2 Commentary Part 2Document110 pagesFema 450 2 Commentary Part 2Tariq AbdulsalamNo ratings yet
- Novelty Yarn 2Document2 pagesNovelty Yarn 2Md Ariful IslamNo ratings yet
- Bio Ethanol DBGDocument30 pagesBio Ethanol DBGGhulam RabbaniNo ratings yet
- Coefficient of Consolidation and Its Correlation With Index Properties of Remolded SoilsDocument6 pagesCoefficient of Consolidation and Its Correlation With Index Properties of Remolded SoilsSanjeev KumarNo ratings yet
- 10.1201 b15488 PreviewpdfDocument157 pages10.1201 b15488 Previewpdflongcongchim123100% (2)
- CMMT61Document78 pagesCMMT61İsmihan AltinNo ratings yet
- 3.7M-Mechanical Analysis of Antenna Structure Design PDFDocument8 pages3.7M-Mechanical Analysis of Antenna Structure Design PDFAbdessamad AmarNo ratings yet
- Cimcool Cimstar 620nfmDocument2 pagesCimcool Cimstar 620nfmtribolasNo ratings yet
- SIPs Frequently Asked QuestionsDocument10 pagesSIPs Frequently Asked QuestionsJoe DinglasanNo ratings yet
- Structural Engineering: Indian Society OF Structural EngineersDocument24 pagesStructural Engineering: Indian Society OF Structural Engineerssuresh_501No ratings yet
- VDZ ActivityReport07 09Document140 pagesVDZ ActivityReport07 09jorge_acosta_112100% (1)
- Chemistry: University of Cambridge International Examinations General Certificate of Education Ordinary LevelDocument16 pagesChemistry: University of Cambridge International Examinations General Certificate of Education Ordinary LevelKelvin SerimweNo ratings yet
- LOK Test PDFDocument4 pagesLOK Test PDFdream4755No ratings yet
- Endogenic Processes Erosion and DepositionDocument8 pagesEndogenic Processes Erosion and Depositionchristan yapNo ratings yet
- Well Workover and InterventionDocument27 pagesWell Workover and InterventionAndrewNo ratings yet