Download as pdf or txt
You might also like
- Cookbook FinalDocument40 pagesCookbook FinalmelissaNo ratings yet
- Problem Solving in RheumatologyDocument293 pagesProblem Solving in RheumatologyGaudeamus IgiturNo ratings yet
- Nephrotic Case Study With AnswersDocument3 pagesNephrotic Case Study With AnswersKBNo ratings yet
- Reproduction in Humans: Test Yourself 18.1 (Page 335)Document4 pagesReproduction in Humans: Test Yourself 18.1 (Page 335)leeNo ratings yet
- Idiopathic Pulmonary Fibrosis: Optimizing The Diagnosis and Multi-Disciplinary Decision MakingDocument4 pagesIdiopathic Pulmonary Fibrosis: Optimizing The Diagnosis and Multi-Disciplinary Decision MakingAnna LiachenkoNo ratings yet
- (13360329 - Endocrine Regulations) Pheochromocytoma, Diagnosis and Treatment - Review of The LiteratureDocument14 pages(13360329 - Endocrine Regulations) Pheochromocytoma, Diagnosis and Treatment - Review of The LiteratureNi Nyoman Indirawati KusumaNo ratings yet
- Idiopathic Pulmonary Fibrosis: Diagnosis, Epidemiology and Natural HistoryDocument11 pagesIdiopathic Pulmonary Fibrosis: Diagnosis, Epidemiology and Natural HistorySuwandi AlghozyNo ratings yet
- Pheochromocytoma Diagnosis and Treatment Review ofDocument14 pagesPheochromocytoma Diagnosis and Treatment Review ofZul FahmiNo ratings yet
- Olivier 2017Document5 pagesOlivier 2017Ivan MihailovicNo ratings yet
- Hermansky-Pudlak Syndrome: Case ReportDocument3 pagesHermansky-Pudlak Syndrome: Case ReportAjanapakhi SahaNo ratings yet
- Fepp Ats 2019Document9 pagesFepp Ats 2019aabigutNo ratings yet
- Fulminant Hepatic FailureDocument12 pagesFulminant Hepatic Failureafghansyah arfiantoNo ratings yet
- International Journal of Infectious DiseasesDocument3 pagesInternational Journal of Infectious DiseasesPendidikan Dokter Unsyiah 2015No ratings yet
- Prophylaxis Against Fungal Peritonitis in CAPD A Single Center Experience With Low Dose FluconazoleDocument5 pagesProphylaxis Against Fungal Peritonitis in CAPD A Single Center Experience With Low Dose FluconazoleJorge David Gamez PeñarandaNo ratings yet
- Derrame Pleural 3Document46 pagesDerrame Pleural 3Héctor David GalánNo ratings yet
- Hypophosphatasia The Disease in AdultsDocument6 pagesHypophosphatasia The Disease in Adultsadrip234No ratings yet
- An Update On Feline Infectious Peritonitis - Diagnostics and TherapeuticsDocument9 pagesAn Update On Feline Infectious Peritonitis - Diagnostics and TherapeuticsRhagaraRhaNo ratings yet
- 34 Iajps34102020Document7 pages34 Iajps34102020iajpsNo ratings yet
- HistoDocument18 pagesHistoHakham DoomguideNo ratings yet
- Infectious Dan Autoantibody Associated EncephalitisDocument13 pagesInfectious Dan Autoantibody Associated EncephalitisAyudiah ParamitaNo ratings yet
- CVJ 10 1091Document3 pagesCVJ 10 1091Rayza LubisNo ratings yet
- J of Small Animal Practice - 2011 - Giori - Performances of Different Diagnostic Tests For Feline Infectious Peritonitis inDocument6 pagesJ of Small Animal Practice - 2011 - Giori - Performances of Different Diagnostic Tests For Feline Infectious Peritonitis inAna DobrinNo ratings yet
- Porphyria The Neglected DiagnosisDocument2 pagesPorphyria The Neglected Diagnosisstudentstoma61No ratings yet
- Incidence and Prevalence of Idiopathic Pulmonary Fibrosis: Review of The LiteratureDocument7 pagesIncidence and Prevalence of Idiopathic Pulmonary Fibrosis: Review of The Literature21-010 AisyahNo ratings yet
- Acute Porpyria Management GuidelineDocument24 pagesAcute Porpyria Management GuidelineGPNNo ratings yet
- EJE Cuny2013Document9 pagesEJE Cuny2013W Antonio Muñoz ChNo ratings yet
- Rigid BronchosDocument6 pagesRigid BronchosVania Dwi AndhaniNo ratings yet
- Bellini2009 PDFDocument8 pagesBellini2009 PDFJohanna Erika Avila FigueroaNo ratings yet
- Origins of Syphilis and Management in The Immunocompetent Patient: Facts and ControversiesDocument6 pagesOrigins of Syphilis and Management in The Immunocompetent Patient: Facts and ControversiesegerpratamaNo ratings yet
- Pediatric Systemic Lupus Erythematosus Associated With AutoimmuneDocument9 pagesPediatric Systemic Lupus Erythematosus Associated With AutoimmunegistaluvikaNo ratings yet
- Can Tuberculous Pleural Effusions Be Diagnosed by Pleural Fluid Analysis Alone?Document8 pagesCan Tuberculous Pleural Effusions Be Diagnosed by Pleural Fluid Analysis Alone?AuliaHadiningratNo ratings yet
- Chary Et Al 2020 Prevalence and Recovery From Olfactory and Gustatory Dysfunctions in Covid 19 Infection A ProspectiveDocument8 pagesChary Et Al 2020 Prevalence and Recovery From Olfactory and Gustatory Dysfunctions in Covid 19 Infection A Prospectiveika nurul shofiyahNo ratings yet
- Revue PPFDocument5 pagesRevue PPFWilliam Del ConteNo ratings yet
- Oropharyngeal Dysphagia After Stroke: Incidence, Diagnosis, and Clinical Predictors in Patients Admitted To A Neurorehabilitation UnitDocument7 pagesOropharyngeal Dysphagia After Stroke: Incidence, Diagnosis, and Clinical Predictors in Patients Admitted To A Neurorehabilitation UnitfaradibaNo ratings yet
- 2005 - Recommendations For DX and TTMT of Acute PorphyriasDocument13 pages2005 - Recommendations For DX and TTMT of Acute PorphyriasDavidArangoNo ratings yet
- Streptococcus Pneumoniae Associated HemolyticDocument11 pagesStreptococcus Pneumoniae Associated HemolyticAnonymous RxCl5asNo ratings yet
- Iron Defi Ciency Anaemia: SeminarDocument10 pagesIron Defi Ciency Anaemia: SeminarShalinee PrasadNo ratings yet
- 1 IiomainDocument6 pages1 IiomainyandaputriNo ratings yet
- Nationwide Prevalence of Sporadic and Familial Idiopathic Pulmonary Fibrosis: Evidence of Founder Effect Among Multiplex Families in FinlandDocument5 pagesNationwide Prevalence of Sporadic and Familial Idiopathic Pulmonary Fibrosis: Evidence of Founder Effect Among Multiplex Families in FinlandNailatul Izza HumammyNo ratings yet
- Heart Failure With Preserved and Reduced EjectionDocument4 pagesHeart Failure With Preserved and Reduced EjectionmirzaNo ratings yet
- Fever of Unknown OriginDocument6 pagesFever of Unknown OriginTio Prima SNo ratings yet
- Atypical Pituitary Adenoma: A Case ReportDocument3 pagesAtypical Pituitary Adenoma: A Case ReportMuhammad Hafizh Islam SadidaNo ratings yet
- Uhm2008 PDFDocument5 pagesUhm2008 PDFTony LeeNo ratings yet
- A Pituitary Mass Is Not Always An Adenoma A Rar - 2024 - International JournalDocument6 pagesA Pituitary Mass Is Not Always An Adenoma A Rar - 2024 - International JournalRonald QuezadaNo ratings yet
- Potter'S Syndrome: A Study of 15 Patients: Original ArticleDocument4 pagesPotter'S Syndrome: A Study of 15 Patients: Original ArticleyanuarizkaNo ratings yet
- 1543-2165-134 6 815 PDFDocument11 pages1543-2165-134 6 815 PDFmmigod666No ratings yet
- Prevalence of Rheumatic Heart Disease Detected by Echocardiographic ScreeningDocument7 pagesPrevalence of Rheumatic Heart Disease Detected by Echocardiographic Screeningandamar0290No ratings yet
- Zhao2017 PDFDocument4 pagesZhao2017 PDFMichelle PhenNo ratings yet
- A Retrospective Analysis of Fourniers Gangrene at A TertiaryGovernment Hospital in The PhilippinesDocument3 pagesA Retrospective Analysis of Fourniers Gangrene at A TertiaryGovernment Hospital in The PhilippinesGian PagadduNo ratings yet
- Leukoplakia or LPR The Misdiagnosis Orf Laryngeal TuberculosisDocument5 pagesLeukoplakia or LPR The Misdiagnosis Orf Laryngeal Tuberculosisharyo wiryantoNo ratings yet
- Upsurge of Cases of Lichen Planus in Iraqi Population in Baghdad City With Frequency of Hepatitis VirusesDocument4 pagesUpsurge of Cases of Lichen Planus in Iraqi Population in Baghdad City With Frequency of Hepatitis VirusesIOSRjournalNo ratings yet
- Prognostic Factors of Death in Leptospirosis: A Prospective Cohort Study in Khon Kaen, ThailandDocument8 pagesPrognostic Factors of Death in Leptospirosis: A Prospective Cohort Study in Khon Kaen, ThailandSilvia RNo ratings yet
- Syphilitic Uveitis - EyeWikiDocument5 pagesSyphilitic Uveitis - EyeWikitgrrwccj98No ratings yet
- Awt 345Document14 pagesAwt 345adinda.larastitiNo ratings yet
- Cord Blood StudyDocument3 pagesCord Blood StudygemaNo ratings yet
- Hepatic Encephalopathy Novel Insights Into Classification, Pathophysiology and TherapyDocument22 pagesHepatic Encephalopathy Novel Insights Into Classification, Pathophysiology and TherapyMichelle RdgzNo ratings yet
- Lupus Nephritis... CureusDocument8 pagesLupus Nephritis... CureusAnab Rehan TaseerNo ratings yet
- 1 s2.0 S003962571630090X MainDocument14 pages1 s2.0 S003962571630090X MainBenjamin NgNo ratings yet
- Patient Handbook BrochureDocument8 pagesPatient Handbook BrochureDr-Yasir Mahmood SyedNo ratings yet
- 2779 12418 1 PBDocument5 pages2779 12418 1 PBChristopher Surya SuwitaNo ratings yet
- Clinical and Genetic Characteristics of Xeroderma Pigmentosum in NepalDocument18 pagesClinical and Genetic Characteristics of Xeroderma Pigmentosum in NepalKandy RinconNo ratings yet
- Article Casey LIVER Hepatic CirrhosisDocument8 pagesArticle Casey LIVER Hepatic CirrhosisMaría José Muñoz PalaciosNo ratings yet
- Uncommon Disorders Masquerading As Acute Flaccid Paralysis in ChildrenDocument7 pagesUncommon Disorders Masquerading As Acute Flaccid Paralysis in ChildrenAnonymous G20oAbl6p8No ratings yet
- Bronsted QA Set C Bio 1 GuideDocument8 pagesBronsted QA Set C Bio 1 GuideBaguma MichaelNo ratings yet
- g6pd Group 5 A2Document39 pagesg6pd Group 5 A2Kristian CadaNo ratings yet
- Non Communicable DiseasesDocument13 pagesNon Communicable Diseaseszzzsubedi100% (1)
- Niu 2024Document10 pagesNiu 2024Gustavo ColodroNo ratings yet
- Metabolic Changes of DrugsDocument103 pagesMetabolic Changes of DrugsDaniel Wang100% (2)
- Vector-Borne Diseases JEdengue Bauko2019Document42 pagesVector-Borne Diseases JEdengue Bauko2019Cyra Oynang100% (1)
- Acceptance Letter CsuDocument3 pagesAcceptance Letter CsuRufaq UllahNo ratings yet
- Lab Manual GeneticsDocument15 pagesLab Manual Geneticsapi-319985329100% (1)
- Cloning Lesson For High School ClassesDocument9 pagesCloning Lesson For High School Classesmartin pontNo ratings yet
- Myopia PresentationDocument18 pagesMyopia PresentationEevaeNo ratings yet
- Ch. 7 DNA Profiling Notes PP GCDocument62 pagesCh. 7 DNA Profiling Notes PP GCrpb barNo ratings yet
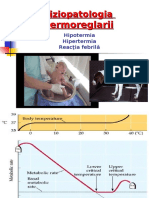
- Documents - MX Fiziopatologia Termoreglarii Fiziopatologia Termoreglarii Hipotermia Hipertermia Reactia Febrila 10-12-2006Document42 pagesDocuments - MX Fiziopatologia Termoreglarii Fiziopatologia Termoreglarii Hipotermia Hipertermia Reactia Febrila 10-12-2006AndreiNo ratings yet
- Specific Language Impairment: A Deficit in Grammar or Processing?Document8 pagesSpecific Language Impairment: A Deficit in Grammar or Processing?Isabel PereiraNo ratings yet
- Short Listed For InterviewDocument34 pagesShort Listed For Interviewathar_tNo ratings yet
- Bacterial TransformationDocument5 pagesBacterial TransformationSubrata KunduNo ratings yet
- Chemistry Class 12 Chemical Kinetics ProjectDocument11 pagesChemistry Class 12 Chemical Kinetics ProjectNich TeniNo ratings yet
- 01 Biology and The Tree of LifeDocument53 pages01 Biology and The Tree of LifeDFurgioneNo ratings yet
- Early Breast Cancer - From Screening To Multidisciplinary Management - 3E (PDF) (UnitedVRG)Document586 pagesEarly Breast Cancer - From Screening To Multidisciplinary Management - 3E (PDF) (UnitedVRG)Luciana Bruno MaiaNo ratings yet
- Bruce Lipton InterviewDocument28 pagesBruce Lipton InterviewDr Ron100% (2)
- Olfactory Nerve - WikipediaDocument29 pagesOlfactory Nerve - Wikipediaراوند اعبيدNo ratings yet
- Kite Corporate Presentation 4-15-15 - Jefferies Immuno-Oncology SummitDocument27 pagesKite Corporate Presentation 4-15-15 - Jefferies Immuno-Oncology SummitJake MannNo ratings yet
- Cloning Tech Guide PDFDocument40 pagesCloning Tech Guide PDFLuis Fernando Soto UgaldiNo ratings yet
- Tuberculinum PDFDocument4 pagesTuberculinum PDFadi swara100% (1)
- Persentase SOAL UKMPPDDocument18 pagesPersentase SOAL UKMPPDFadhal Muhammad AhmadNo ratings yet
- BIOMATERIALSDocument3 pagesBIOMATERIALSrajarshidharNo ratings yet
- Khusbu Keyal 2017 PDFDocument10 pagesKhusbu Keyal 2017 PDFFelicia SutarliNo ratings yet