Download as pdf or txt
You might also like
- Sucessful Business Intelligence PDFDocument337 pagesSucessful Business Intelligence PDFNikita GoncharNo ratings yet
- Globin Gene & Molecular Clock PDFDocument36 pagesGlobin Gene & Molecular Clock PDFManisha BishtNo ratings yet
- Chapter 3c X Ray DiffractionDocument48 pagesChapter 3c X Ray DiffractionAnup DalalNo ratings yet
- Lecture 1 Principle and Application of X Ray DiffractometerDocument39 pagesLecture 1 Principle and Application of X Ray DiffractometerVadivelanNo ratings yet
- 8 X-Ray CrystallDocument12 pages8 X-Ray CrystallGustavo AraújoNo ratings yet
- Https - Myguru - Upsi.edu - My - Documents - 2019 - Courses - SFT3053 - Material - K00926 - 20191001220515 - Chapter 2 FLS PDFDocument46 pagesHttps - Myguru - Upsi.edu - My - Documents - 2019 - Courses - SFT3053 - Material - K00926 - 20191001220515 - Chapter 2 FLS PDFNurazin RizalNo ratings yet
- 10 X-Ray DiffractionDocument8 pages10 X-Ray DiffractionProf.Dr.Mohamed Fahmy Mohamed Hussein100% (1)
- MSE8013 Chapter02 Wave Diffraction and The Reciprocal LatticeDocument61 pagesMSE8013 Chapter02 Wave Diffraction and The Reciprocal LatticeJie GanNo ratings yet
- XRDDocument27 pagesXRDBaraliya Jagdish DNo ratings yet
- X-Ray Crystallography X-RayDocument10 pagesX-Ray Crystallography X-RayJoriel SolenonNo ratings yet
- X-Ray Diffraction (XRD) : Ulrike TroitzschDocument38 pagesX-Ray Diffraction (XRD) : Ulrike TroitzschprianNo ratings yet
- X-Ray Diffraction Basic Concepts - DemostrationDocument21 pagesX-Ray Diffraction Basic Concepts - DemostrationAhmed MaatyNo ratings yet
- Kimia Analisis Instrumen 2: X-Ray DiffractionDocument28 pagesKimia Analisis Instrumen 2: X-Ray Diffractionsofi salasiNo ratings yet
- Structure Determination Through Z-Contrast Microscopy: S. J. PennycookDocument32 pagesStructure Determination Through Z-Contrast Microscopy: S. J. PennycookevermorebattleNo ratings yet
- XRD PPT Part 2Document39 pagesXRD PPT Part 2BME62Thejeswar SeggamNo ratings yet
- X-Ray: Radiation, After The German Scientist Wilhelm Röntgen, WhoDocument16 pagesX-Ray: Radiation, After The German Scientist Wilhelm Röntgen, WhoJames MoriiartyNo ratings yet
- X-Ray Diffraction (XRD)Document31 pagesX-Ray Diffraction (XRD)Yulianto NugrohoNo ratings yet
- 8 XRDDocument14 pages8 XRDWahyu Eko PrasetyoNo ratings yet
- Atoms Solids Crystalline:) Transmission Electron MicroscopeDocument6 pagesAtoms Solids Crystalline:) Transmission Electron MicroscopeAtul DwivediNo ratings yet
- Reference Book: Elements of X-Ray DiffractionDocument14 pagesReference Book: Elements of X-Ray Diffractiondtu epNo ratings yet
- Chapter 3c X Ray DiffractionDocument51 pagesChapter 3c X Ray DiffractionDeependra Kumar Ban100% (1)
- Chapter 3c X Ray DiffractionDocument51 pagesChapter 3c X Ray Diffractionkaushaltrivedi46No ratings yet
- De Broglie's Equation:: Electron DiffractionDocument6 pagesDe Broglie's Equation:: Electron DiffractionSaleem KhanNo ratings yet
- Lecture 06Document32 pagesLecture 06Retro lashNo ratings yet
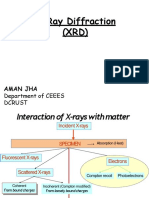
- X Ray DiffractionsDocument15 pagesX Ray DiffractionsAman JhaNo ratings yet
- Seminar On X-Ray Crystallography: Presented by Mounik Rout M.Pharm (PH - Technology) Guided by Dr. Sasmita Kumari AcharjyaDocument31 pagesSeminar On X-Ray Crystallography: Presented by Mounik Rout M.Pharm (PH - Technology) Guided by Dr. Sasmita Kumari AcharjyaMounik RoutNo ratings yet
- Basic X-Ray Powder Diffraction (XRPD) : In-Situ ExperimentsDocument47 pagesBasic X-Ray Powder Diffraction (XRPD) : In-Situ ExperimentsImran KhanNo ratings yet
- Determination of Crystal Structure and Crystallite SizeDocument17 pagesDetermination of Crystal Structure and Crystallite SizeRohit SatheshNo ratings yet
- Chapter 3c X Ray DiffractionDocument48 pagesChapter 3c X Ray DiffractionmethoxyNo ratings yet
- X-ray Diffraction:: X-rays are electromagnetic waves (λ = 0.1 - 100 Å)Document6 pagesX-ray Diffraction:: X-rays are electromagnetic waves (λ = 0.1 - 100 Å)Miles NsgNo ratings yet
- Structure Determination Through Z-Contrast Microscopy: S. J. PennycookDocument32 pagesStructure Determination Through Z-Contrast Microscopy: S. J. Pennycookpiratina33No ratings yet
- Characterization of NanomaterialsDocument75 pagesCharacterization of NanomaterialsAbhishek SharmaNo ratings yet
- HAFTA XRD-1-dönüştürüldüDocument26 pagesHAFTA XRD-1-dönüştürüldüKatrina Ranee RamosNo ratings yet
- X RaysDocument11 pagesX RaysCSF1No ratings yet
- Mod Ch3matDocument15 pagesMod Ch3matJulian David Henao EscobarNo ratings yet
- LaserDocument23 pagesLaserAnkit UpadhyayNo ratings yet
- 5-X Ray DiffractionDocument24 pages5-X Ray DiffractionYagnesh Rohit100% (1)
- Electron Probe Microanalysis Epma: Related Topics: X-Ray Fluorescence (XRF) and Synchrotron RadiationDocument35 pagesElectron Probe Microanalysis Epma: Related Topics: X-Ray Fluorescence (XRF) and Synchrotron RadiationAmit SawNo ratings yet
- Spectroscopic Techniques - XRDDocument18 pagesSpectroscopic Techniques - XRDRitik raj mehraNo ratings yet
- Interaction of X RaysDocument50 pagesInteraction of X RaysAzharNo ratings yet
- Chapter 3c X Ray DiffractionDocument40 pagesChapter 3c X Ray DiffractiondhandametNo ratings yet
- X-Ray Diffraction 7Document11 pagesX-Ray Diffraction 7alina.tlekkabylova270202No ratings yet
- X - Ray Diffraction (XRD)Document26 pagesX - Ray Diffraction (XRD)Ajith KumarNo ratings yet
- 411 SydDocument159 pages411 SydUtsho DasNo ratings yet
- Solid State Chapter 2Document75 pagesSolid State Chapter 2Ambreen KhanNo ratings yet
- X RayDocument8 pagesX RayamehnrNo ratings yet
- X-Ray Diffraction Analysis Principle InsDocument57 pagesX-Ray Diffraction Analysis Principle InsPrem Kumar100% (1)
- Medical PhysicsDocument49 pagesMedical PhysicsKeanan WongsoNo ratings yet
- XRD (X - Ray Diffraction)Document7 pagesXRD (X - Ray Diffraction)summi64No ratings yet
- Lecture - Electron DiffractionDocument12 pagesLecture - Electron DiffractionOlivia WahyudiNo ratings yet
- Unit 4 Structure Unfolding TechniquesDocument29 pagesUnit 4 Structure Unfolding TechniquesvijayNo ratings yet
- Electron Microscopy BasicsDocument45 pagesElectron Microscopy BasicsNithiesh NaikNo ratings yet
- Lecture 10 - 0Document69 pagesLecture 10 - 0yhjklNo ratings yet
- Xray Diffraction ReportDocument30 pagesXray Diffraction ReportBrian Hallee100% (1)
- Fundamentals X-Ray DiffractionDocument14 pagesFundamentals X-Ray DiffractionArif MamonNo ratings yet
- X Ray Diffraction PDFDocument9 pagesX Ray Diffraction PDFYousef Adel HassanenNo ratings yet
- Slides MLL100 5th WeekDocument69 pagesSlides MLL100 5th WeekTanishka GurjarNo ratings yet
- Basics of X-Ray DiffractionDocument13 pagesBasics of X-Ray Diffractionleizar_death64No ratings yet
- Part 1. WRICM PDFDocument22 pagesPart 1. WRICM PDFPatrick PascasioNo ratings yet
- EFuel100 Product Information BrochureDocument2 pagesEFuel100 Product Information BrochurekuttraNo ratings yet
- Suzuki Ertiga p1-26Document26 pagesSuzuki Ertiga p1-26bonruiz100% (2)
- Variable Valve Timing & Acoustic Control Induction Systems: Section 8Document19 pagesVariable Valve Timing & Acoustic Control Induction Systems: Section 8sungjoo75No ratings yet
- RESUN TUV-CE Certificate 20220919Document2 pagesRESUN TUV-CE Certificate 20220919gigatech.roNo ratings yet
- RT Usersguide PDFDocument36 pagesRT Usersguide PDFRoy RamosNo ratings yet
- Tornillo Autop. P-BR ZN 10 X 3-4 (Pat-21138 + T)Document1 pageTornillo Autop. P-BR ZN 10 X 3-4 (Pat-21138 + T)L Caqui PascacioNo ratings yet
- CV - Emad Jamshidi: P Date of Birth Gender Citizenship E-Mail Phone Work Mobile Telephone Current ProfessionDocument1 pageCV - Emad Jamshidi: P Date of Birth Gender Citizenship E-Mail Phone Work Mobile Telephone Current ProfessionEmad JamshidiNo ratings yet
- CH - 4 - THE RATTRAP - pdf1684425205-smsDocument4 pagesCH - 4 - THE RATTRAP - pdf1684425205-smsAnanya SamantaNo ratings yet
- Technical Offer Sama Baghdad Company 31st DEC 2020 Solicitation W56KGZ21Q7017Document120 pagesTechnical Offer Sama Baghdad Company 31st DEC 2020 Solicitation W56KGZ21Q7017Ra'ad HaniNo ratings yet
- Turbo HD DVR V3.4.2 Release Notes - ExternalDocument3 pagesTurbo HD DVR V3.4.2 Release Notes - Externalcrishtopher saenzNo ratings yet
- A Simplified Method For Assessing The Saturation Efficiency at Full-Scale Dissolved Air Flotation Plant PDFDocument8 pagesA Simplified Method For Assessing The Saturation Efficiency at Full-Scale Dissolved Air Flotation Plant PDFChris QueroNo ratings yet
- Soft Ferrite Cores User Guide PDFDocument52 pagesSoft Ferrite Cores User Guide PDFPrithvi ScorpNo ratings yet
- Determination of Pack-Set Index of Portland Cement: Standard Test Method ForDocument5 pagesDetermination of Pack-Set Index of Portland Cement: Standard Test Method ForAhmed AlzubaidiNo ratings yet
- CEM130 Construction Safety Management - SYLLABUS RevisedDocument5 pagesCEM130 Construction Safety Management - SYLLABUS Reviseddel rosarioNo ratings yet
- Senior HighschoolDocument3 pagesSenior HighschoolAngelica MathewNo ratings yet
- English TestDocument5 pagesEnglish TestfabiolaNo ratings yet
- RSL Cbta Plan: (KVB) (Teknologi Elektronik Etn 102)Document3 pagesRSL Cbta Plan: (KVB) (Teknologi Elektronik Etn 102)Ha Lim HasanNo ratings yet
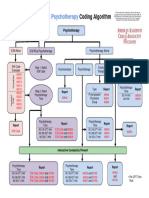
- E and M and Psychotherapy Coding AlgorithmDocument1 pageE and M and Psychotherapy Coding AlgorithmRashiden MadjalesNo ratings yet
- The Modernist Moment at The University of LeedsDocument26 pagesThe Modernist Moment at The University of LeedstobyNo ratings yet
- MapmyIndia Google Distance MatrixDocument3 pagesMapmyIndia Google Distance Matrixrama.j266No ratings yet
- Saha, 2003Document16 pagesSaha, 2003LA ArenqueNo ratings yet
- Real Time Characteristic Value CombinationDocument5 pagesReal Time Characteristic Value CombinationHari KrishnaNo ratings yet
- Disability Tax Credit InstructionsDocument12 pagesDisability Tax Credit InstructionsShauna PaynterNo ratings yet
- J6-S9014 DatasheetDocument2 pagesJ6-S9014 DatasheetOreol100% (1)
- Scada & HmiDocument57 pagesScada & HmiwronghNo ratings yet
- Considerations in Contact Lens Use Under Adverse ConditionsDocument178 pagesConsiderations in Contact Lens Use Under Adverse ConditionsGusti Andhika AzwarNo ratings yet
- Design and Fabrication of Mini-Air EngineDocument33 pagesDesign and Fabrication of Mini-Air EngineMadhan KumarNo ratings yet