Download as pdf or txt
You might also like
- STP 767Document13 pagesSTP 767vatrushka69No ratings yet
- Lab Report Distillation Column PDFDocument26 pagesLab Report Distillation Column PDFGebrina RizkiaNo ratings yet
- Lab 6 - Cellular Respiration - The Citric Acid Cycle ReactionsDocument9 pagesLab 6 - Cellular Respiration - The Citric Acid Cycle ReactionsDan Pingel100% (1)
- Deep FreezerDocument15 pagesDeep FreezerAbhishek MahajanNo ratings yet
- Jimmaa University College of Natural Sciences Department of ChemistryDocument14 pagesJimmaa University College of Natural Sciences Department of ChemistrywoldNo ratings yet
- Supercritical Fluid Extraction: by Nicole Adams and Morgan CampbellDocument27 pagesSupercritical Fluid Extraction: by Nicole Adams and Morgan CampbellAni KushwahaNo ratings yet
- Sample Prep NoteDocument89 pagesSample Prep NotesyokriNo ratings yet
- Organic Chemistry: Basra University College of Science and Technology Pharmacy DepartmentDocument10 pagesOrganic Chemistry: Basra University College of Science and Technology Pharmacy DepartmentcrtgyhujikNo ratings yet
- GC AnalysisDocument5 pagesGC Analysisabul1148No ratings yet
- Cleaning Validation Practices Using A Single Pot ProcessorDocument9 pagesCleaning Validation Practices Using A Single Pot Processorjljimenez1969No ratings yet
- DryingDocument25 pagesDryingVeena Subramanian0% (1)
- Oil Seed Analysis Lab ReportDocument13 pagesOil Seed Analysis Lab ReportMohamad Samer KansouNo ratings yet
- Phytochemistry: Authentication of The PlantDocument10 pagesPhytochemistry: Authentication of The PlantFeras SwaidNo ratings yet
- Methods of ExtractionDocument39 pagesMethods of ExtractionVijetha PendyalaNo ratings yet
- BR Rocket Evaporator BR70315 EDocument8 pagesBR Rocket Evaporator BR70315 EAndrés MárquezNo ratings yet
- Chap-1-2 ExtractionDocument73 pagesChap-1-2 Extractionlishan asefaNo ratings yet
- LleDocument30 pagesLlefirstlove_492_736373No ratings yet
- Vapor DepositionDocument7 pagesVapor DepositionDebabrata DeyNo ratings yet
- ExtractionDocument65 pagesExtractionmanchandaraghav05No ratings yet
- EXTRACTION Microwave-Assisted ExtractionDocument10 pagesEXTRACTION Microwave-Assisted Extractionerlinda rhohmatul lailiNo ratings yet
- Case 01Document19 pagesCase 01Rushanth ChandraboseNo ratings yet
- Packed Bed Distillation ColumnDocument20 pagesPacked Bed Distillation ColumnAmoluck BhatiaNo ratings yet
- SCH 410 Lecture One 2023Document20 pagesSCH 410 Lecture One 2023Samuel MutisyaNo ratings yet
- Extraction of Chlorophyll From Alfalfa PlantDocument13 pagesExtraction of Chlorophyll From Alfalfa PlantAhmed AliNo ratings yet
- Department of Pharmacy: The Extraction MR - Ahmed AlmohhmmedDocument10 pagesDepartment of Pharmacy: The Extraction MR - Ahmed AlmohhmmedcrtgyhujikNo ratings yet
- VI Sem Mass Transfer Lab ManualDocument53 pagesVI Sem Mass Transfer Lab Manualoctoviancletus80% (10)
- MIC455 AssignmentDocument13 pagesMIC455 AssignmentFARIHA ALAMNo ratings yet
- BY-Prince Mishra B.Voc. FT5Document19 pagesBY-Prince Mishra B.Voc. FT5Giri PrasadNo ratings yet
- Liquid LiquidDocument8 pagesLiquid LiquidAnonymous b9fcR5No ratings yet
- CHEM 1015 Lab 2 - ExtractionDocument17 pagesCHEM 1015 Lab 2 - ExtractionKnzy ElmasryNo ratings yet
- Hill Reaction of Photosynthesis-Effects of Selected HerbicidesDocument4 pagesHill Reaction of Photosynthesis-Effects of Selected HerbicidesBryan AbarcaNo ratings yet
- Proposed Models For Subcritical Water Extraction of Essential OilsDocument7 pagesProposed Models For Subcritical Water Extraction of Essential OilsTamadur BarghoothiNo ratings yet
- Lab Report (Environmental)Document38 pagesLab Report (Environmental)Muhammad ShahbazNo ratings yet
- Continuous Stirred Tank ReactorDocument7 pagesContinuous Stirred Tank ReactordeepshikhasinghNo ratings yet
- Mass Transfer Lab ManuAL - 2Document18 pagesMass Transfer Lab ManuAL - 2VigneshParthasarathy0% (1)
- Distillation Lab Manual PDFDocument12 pagesDistillation Lab Manual PDFIdil DoreNo ratings yet
- Chapter 7.2Document20 pagesChapter 7.2Aakash MandalNo ratings yet
- 5.1 VOC Determination COV According To The Method SR EN ISO 16017/1-2003Document4 pages5.1 VOC Determination COV According To The Method SR EN ISO 16017/1-2003Renata BrudascaNo ratings yet
- Supercritical Fluid ExtractionDocument5 pagesSupercritical Fluid ExtractionUsman WaheedNo ratings yet
- Components of A FermenterDocument7 pagesComponents of A FermenterVarun Rajput0% (1)
- Experiment Manual - Liquid-Liquid ExtractionDocument24 pagesExperiment Manual - Liquid-Liquid ExtractionStolen RememberNo ratings yet
- ExtractionDocument5 pagesExtractionRiya VishwakarmaNo ratings yet
- Different Method's of ExtractionDocument10 pagesDifferent Method's of ExtractionVikas VikiNo ratings yet
- Extraction of Insulin Like Compound From Bitter MelonDocument35 pagesExtraction of Insulin Like Compound From Bitter MelonNurul Hidayah Hamzah100% (1)
- What Is Gas Chromatography AssignmentDocument10 pagesWhat Is Gas Chromatography AssignmentIsmi Fadli100% (1)
- تقريرDocument8 pagesتقريرAbduallah Thaer HassanNo ratings yet
- Recovery of Bio-ProductsDocument32 pagesRecovery of Bio-ProductsH.J.Prabhu67% (3)
- Report 5 - OC LabDocument10 pagesReport 5 - OC LabBùi Ngọc MaiNo ratings yet
- Env Engg Lab ReportDocument46 pagesEnv Engg Lab ReportMuhammad ShahbazNo ratings yet
- Botany Lec.4Document5 pagesBotany Lec.4ُEl SyrianoNo ratings yet
- Physiology Lab 1 2Document11 pagesPhysiology Lab 1 2tmqt2fbnzgNo ratings yet
- 5 Distillation Final ReportDocument7 pages5 Distillation Final ReportElzubair EljaaliNo ratings yet
- Liquid Liquid ExtractionDocument25 pagesLiquid Liquid ExtractionAllensius Karelsta HarefaNo ratings yet
- Pressurized Liquid Extraction of Avonoids From Houttuynia Cordata ThunbDocument6 pagesPressurized Liquid Extraction of Avonoids From Houttuynia Cordata Thunbnapkato100% (1)
- Module 10Document11 pagesModule 10Syafri LushainiNo ratings yet
- Solvent Extraction Lab ReportDocument11 pagesSolvent Extraction Lab ReportWan Nurshahira100% (2)
- Aykan07.05.2015 20.27.26ders PDFDocument8 pagesAykan07.05.2015 20.27.26ders PDFRobiel GashuNo ratings yet
- Leclaire Rev1Document6 pagesLeclaire Rev1mackerelfishNo ratings yet
- Lab Report-11: Environmental Chemistry (ENE-213) Course Instructor: Dr. Sofia BaigDocument7 pagesLab Report-11: Environmental Chemistry (ENE-213) Course Instructor: Dr. Sofia BaigHaniya SiddiqueNo ratings yet
- LABORATORY MANUAL FOR A MINI PROJECT: MSCB 1113 BIOCHEMISTRY & MICROBIAL PHYSIOLOGYFrom EverandLABORATORY MANUAL FOR A MINI PROJECT: MSCB 1113 BIOCHEMISTRY & MICROBIAL PHYSIOLOGYNo ratings yet
- Asteroid Mining (Project Eram)Document48 pagesAsteroid Mining (Project Eram)VikasNo ratings yet
- Earth Science Module 9-10Document6 pagesEarth Science Module 9-10Lopez AeraNo ratings yet
- For 12th Class Physics :practice Test On Electrostatic FIELD, POTENTIAL & ENERGY@vireshDocument3 pagesFor 12th Class Physics :practice Test On Electrostatic FIELD, POTENTIAL & ENERGY@vireshViresh SinghNo ratings yet
- Characteristics of Power Generation UnitsDocument32 pagesCharacteristics of Power Generation UnitsOMKAR PATILNo ratings yet
- Thomas Dankowski Fisher Intelligence 5thedDocument48 pagesThomas Dankowski Fisher Intelligence 5thedjelissaNo ratings yet
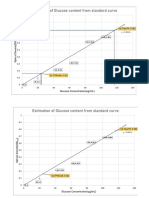
- Estimation of Glucose Content From Standard Curve: Glucose Concentration ( G/ML)Document1 pageEstimation of Glucose Content From Standard Curve: Glucose Concentration ( G/ML)Hasan AnandaNo ratings yet
- Ruta de Síntesis CiprofloxacinaDocument5 pagesRuta de Síntesis CiprofloxacinaJuan VillamilNo ratings yet
- Practice 7Document3 pagesPractice 7Neha SharmaNo ratings yet
- Workbook 7 Answers Cambridge ExtrasDocument28 pagesWorkbook 7 Answers Cambridge ExtrasNithya G100% (1)
- DuPont Non-Stick Dry Film Lubricant - Aerosol - US SDS - 17 Jan 2021Document7 pagesDuPont Non-Stick Dry Film Lubricant - Aerosol - US SDS - 17 Jan 2021Apol Aguisanda JrNo ratings yet
- HMF and Diastase Activity in HoneysDocument30 pagesHMF and Diastase Activity in HoneysТетяна КозицькаNo ratings yet
- Physics 2nd Year - Objective Mcqs - Ali Raza - ArkDocument57 pagesPhysics 2nd Year - Objective Mcqs - Ali Raza - ArkShafia Batool67% (3)
- Exploration of Crude PetroleumDocument13 pagesExploration of Crude PetroleumUsman IsrarNo ratings yet
- 1 s2.0 S026382231730209X MainDocument17 pages1 s2.0 S026382231730209X Mainziwen liuNo ratings yet
- Eddy Current Examination of Steel Tubular Products Using Magnetic SaturationDocument2 pagesEddy Current Examination of Steel Tubular Products Using Magnetic SaturationChenjie ZhuNo ratings yet
- ExoTerra Data Sheet Thrusters REV 0JDocument2 pagesExoTerra Data Sheet Thrusters REV 0Jsghscribd2012No ratings yet
- CEA Report On Coal BlendingDocument50 pagesCEA Report On Coal BlendingSudhir Jadhav100% (1)
- C2 IPL - Chemistry Year Plans 22-23Document8 pagesC2 IPL - Chemistry Year Plans 22-23aadhithyac3iplgNo ratings yet
- Is 4432.1988 PDFDocument15 pagesIs 4432.1988 PDFNav TalukdarNo ratings yet
- 0625 s07 QP 1Document20 pages0625 s07 QP 1Hubbak Khan0% (1)
- CBSE Sample Paper For Class 6 ScienceDocument7 pagesCBSE Sample Paper For Class 6 ScienceBhavana ShindeNo ratings yet
- Environmental Product Declaration: International Interzone 954 (Part A & Part B)Document9 pagesEnvironmental Product Declaration: International Interzone 954 (Part A & Part B)maNo ratings yet
- Topic403 (Doppler Effect)Document14 pagesTopic403 (Doppler Effect)Jeffry RamonidaNo ratings yet
- Technology Profile EphedrineDocument3 pagesTechnology Profile EphedrineKaran VetNo ratings yet
- Application of Cleaning AgentsDocument29 pagesApplication of Cleaning AgentsRogielyn AscañoNo ratings yet
- Physics Form 4 Notes PDFDocument16 pagesPhysics Form 4 Notes PDFIbrahim hasanNo ratings yet
- Poster On Mendeleev's Periodic Table: Merits LimitationsDocument1 pagePoster On Mendeleev's Periodic Table: Merits LimitationsHemanthNo ratings yet
- GR. - 6 - 3rd Q. - Science - STDocument6 pagesGR. - 6 - 3rd Q. - Science - STcarrel inocentesNo ratings yet
- Cambridge Revision Topic 11.3 and 21.1 With AnswersDocument13 pagesCambridge Revision Topic 11.3 and 21.1 With AnswersMarin PesicNo ratings yet