
Optimized Gene Classification Using Support Vector Machine With Convolutional Neural Network For Cancer Detection From Gene Expression Microarray Data
Optimized Gene Classification Using Support Vector Machine With Convolutional Neural Network For Cancer Detection From Gene Expression Microarray Data
You might also like
- Graham Giller Wilmott TalkDocument31 pagesGraham Giller Wilmott TalkGraham GillerNo ratings yet
- Cs 229, Autumn 2016 Problem Set #2: Naive Bayes, SVMS, and TheoryDocument20 pagesCs 229, Autumn 2016 Problem Set #2: Naive Bayes, SVMS, and TheoryZeeshan Ali SayyedNo ratings yet
- Gene Selection and Classification of Microarray Data Using Convolutional Neural NetworkDocument6 pagesGene Selection and Classification of Microarray Data Using Convolutional Neural NetworkTajbia HossainNo ratings yet
- Cancer Detection Using Convolutional Neural Network: February 2021Document10 pagesCancer Detection Using Convolutional Neural Network: February 2021worldworld312No ratings yet
- Almugren, Alshamlan - 2019 - A Survey On Hybrid Feature Selection Methods in Microarray Gene Expression Data For Cancer ClassificationDocument16 pagesAlmugren, Alshamlan - 2019 - A Survey On Hybrid Feature Selection Methods in Microarray Gene Expression Data For Cancer Classificationiyousafzai1No ratings yet
- Hybrid Model For Detection of Brain Tumor Using Convolution Neural NetworksDocument7 pagesHybrid Model For Detection of Brain Tumor Using Convolution Neural NetworksCSIT iaesprimeNo ratings yet
- A Microarray Gene Expression Data Classification UDocument14 pagesA Microarray Gene Expression Data Classification UHuseyin OztoprakNo ratings yet
- Enhanced Network Anomaly Detection Based On Deep Neural NetworksDocument16 pagesEnhanced Network Anomaly Detection Based On Deep Neural NetworksYishak TadeleNo ratings yet
- Brain Tumour Detection Using M-IRO-Journals-3 4 5Document12 pagesBrain Tumour Detection Using M-IRO-Journals-3 4 5MR. AJAYNo ratings yet
- Performance Analysis and Evaluation of Different Data Mining Algorithms Used For Cancer ClassificationDocument7 pagesPerformance Analysis and Evaluation of Different Data Mining Algorithms Used For Cancer ClassificationPAUL AKAMPURIRANo ratings yet
- Irjet V10i214Document3 pagesIrjet V10i214tanzimreadsNo ratings yet
- Astuti 2018 J. Phys. Conf. Ser. 971 012003Document8 pagesAstuti 2018 J. Phys. Conf. Ser. 971 012003fauzieahmad526No ratings yet
- EtextpathshaalaDocument21 pagesEtextpathshaalaMIGUELNo ratings yet
- 1 s2.0 S0079610722000803 MainDocument13 pages1 s2.0 S0079610722000803 MainsznistvanNo ratings yet
- CRISP-MED-DM A Methodology of Diagnosing Breast CancerDocument13 pagesCRISP-MED-DM A Methodology of Diagnosing Breast CancerInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Genes PaperDocument11 pagesGenes PaperTomas PetittiNo ratings yet
- Comparation Analysis of Ensemble Technique With Boosting (Xgboost) and Bagging (Randomforest) For Classify Splice Junction Dna Sequence CategoryDocument10 pagesComparation Analysis of Ensemble Technique With Boosting (Xgboost) and Bagging (Randomforest) For Classify Splice Junction Dna Sequence CategoryFatrinaNo ratings yet
- FsNET FEature Selection Network On HDDDocument14 pagesFsNET FEature Selection Network On HDDnfbeusebio3962No ratings yet
- Diagnosis of Chronic Brain Syndrome Using Deep LearningDocument10 pagesDiagnosis of Chronic Brain Syndrome Using Deep LearningIJRASETPublicationsNo ratings yet
- Microarray Time SeriesDocument19 pagesMicroarray Time SeriesMohamed MahmoudNo ratings yet
- Efficacy of Algorithms in Deep Learning On Brain Tumor Cancer Detection (Topic Area Deep Learning)Document8 pagesEfficacy of Algorithms in Deep Learning On Brain Tumor Cancer Detection (Topic Area Deep Learning)International Journal of Innovative Science and Research TechnologyNo ratings yet
- Hybrid Genetic Algorithm-Neural Network: Feature Extraction For Unpreprocessed Microarray DataDocument10 pagesHybrid Genetic Algorithm-Neural Network: Feature Extraction For Unpreprocessed Microarray DataTajbia HossainNo ratings yet
- Resolving Gene Expression Data Using Multiobjective Optimization ApproachDocument7 pagesResolving Gene Expression Data Using Multiobjective Optimization ApproachIJIRSTNo ratings yet
- Grid Search-Based Hyperparameter Tuning and Classification of Microarray Cancer DataDocument8 pagesGrid Search-Based Hyperparameter Tuning and Classification of Microarray Cancer DataannalystatNo ratings yet
- Malaria Detection Using Deep Convolutional Neural Networks: ISSN-2394-5125Document9 pagesMalaria Detection Using Deep Convolutional Neural Networks: ISSN-2394-5125GEORGENo ratings yet
- Spaper 4Document11 pagesSpaper 4Rozita JackNo ratings yet
- Review Article: A Review of Feature Selection and Feature Extraction Methods Applied On Microarray DataDocument14 pagesReview Article: A Review of Feature Selection and Feature Extraction Methods Applied On Microarray Datafahad darNo ratings yet
- Microarray Data Analysis and Mining Tools: Bioinformation April 2011Document6 pagesMicroarray Data Analysis and Mining Tools: Bioinformation April 2011Dr. Megha PUNo ratings yet
- Applications of Machine Learning Techniques To Predict Diagnostic Breast CancerDocument11 pagesApplications of Machine Learning Techniques To Predict Diagnostic Breast CancerAkashi DogeyNo ratings yet
- Classification of Cancerous Profiles Using Machine LearningDocument38 pagesClassification of Cancerous Profiles Using Machine LearningRuby IniyaNo ratings yet
- A Study On Deep Learning For Bioinformatics: Panchami - VU, Dr. Hariharan.N, Dr. Manish - TIDocument5 pagesA Study On Deep Learning For Bioinformatics: Panchami - VU, Dr. Hariharan.N, Dr. Manish - TIRahul SharmaNo ratings yet
- Pattern Recognition: V. Bolo N-Canedo, N. Sa Nchez-Maron O, A. Alonso-BetanzosDocument9 pagesPattern Recognition: V. Bolo N-Canedo, N. Sa Nchez-Maron O, A. Alonso-BetanzosTajbia HossainNo ratings yet
- Gene Mutation PDFDocument5 pagesGene Mutation PDFChandan NaiduNo ratings yet
- Research Paper 1 PublicationDocument4 pagesResearch Paper 1 PublicationSri Vidya MakkapatiNo ratings yet
- TS PPT (Autosaved)Document16 pagesTS PPT (Autosaved)Vasiha Fathima RNo ratings yet
- Evolutionary Computational Algorithm by Blending of PPCA and EP-Enhanced Supervised Classifier For Microarray Gene Expression DataDocument10 pagesEvolutionary Computational Algorithm by Blending of PPCA and EP-Enhanced Supervised Classifier For Microarray Gene Expression DataIAES IJAINo ratings yet
- Optimizing Classification Efficiency With Machine Learning Techniques For Pattern MatchingDocument18 pagesOptimizing Classification Efficiency With Machine Learning Techniques For Pattern Matchingm.albaiti111No ratings yet
- 22-GWO Papers-27-02-2024Document16 pages22-GWO Papers-27-02-2024gunjanbhartia2003No ratings yet
- 173 701 1 PBDocument12 pages173 701 1 PBArendraNo ratings yet
- Analytical Study of Diagnosis For Angioplasty and Stents Patients Using Improved Classification TechniqueDocument4 pagesAnalytical Study of Diagnosis For Angioplasty and Stents Patients Using Improved Classification TechniqueEditor IJTSRDNo ratings yet
- Deep Learning Based Convolutional Neural Networks (DLCNN) On Classification Algorithm To Detect The Brain Turnor Diseases Using MRI and CT Scan ImagesDocument8 pagesDeep Learning Based Convolutional Neural Networks (DLCNN) On Classification Algorithm To Detect The Brain Turnor Diseases Using MRI and CT Scan ImagesInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Study On Malaria Detection Using Transfer LearningDocument9 pagesStudy On Malaria Detection Using Transfer LearningIJRASETPublicationsNo ratings yet
- Convolutional Neural Networks Based Plant Leaf Diseases Detection SchemeDocument7 pagesConvolutional Neural Networks Based Plant Leaf Diseases Detection SchemeSudhaNo ratings yet
- A Modified Resnet (MRestNet) Based Classification For Brain Tumor DatasetDocument18 pagesA Modified Resnet (MRestNet) Based Classification For Brain Tumor DatasetVinayaga MoorthyNo ratings yet
- Scmra: A Robust Deep Learning Method To Annotate Scrna-Seq Data With Multiple Reference DatasetsDocument15 pagesScmra: A Robust Deep Learning Method To Annotate Scrna-Seq Data With Multiple Reference DatasetsNavid ArefanNo ratings yet
- Analytical Estimation of Quantum Convolutional Neural Network and Convolutional Neural Network For Breast Cancer DetectionDocument11 pagesAnalytical Estimation of Quantum Convolutional Neural Network and Convolutional Neural Network For Breast Cancer DetectionInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- draftDNAPressChapter v8Document77 pagesdraftDNAPressChapter v8animboyNo ratings yet
- Deep Convolutional Neural Network Framework With Multi-Modal Fusion For Alzheimer's DetectionDocument13 pagesDeep Convolutional Neural Network Framework With Multi-Modal Fusion For Alzheimer's DetectionIJRES teamNo ratings yet
- Jurnal 3Document15 pagesJurnal 3fikaadillah3No ratings yet
- A Deep Neural Network Model Using Random Forest ToDocument9 pagesA Deep Neural Network Model Using Random Forest ToGisselyNo ratings yet
- A Survey in DNA Sequence Classification by Soft Computing MethodsDocument6 pagesA Survey in DNA Sequence Classification by Soft Computing MethodsBackiya Lakshmi KumaresanNo ratings yet
- A Comparative Study of Tuberculosis DetectionDocument5 pagesA Comparative Study of Tuberculosis DetectionMehera Binte MizanNo ratings yet
- A Comparative Study For Brain Tumor Detection Analysis Using CNN and VGG-16 and Its ApplicationDocument8 pagesA Comparative Study For Brain Tumor Detection Analysis Using CNN and VGG-16 and Its ApplicationInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Bio Report ElDocument8 pagesBio Report ElJateen RathodNo ratings yet
- Skin Cancer Classification Using Convolutional Neural NetworksDocument8 pagesSkin Cancer Classification Using Convolutional Neural NetworksNirmalendu KumarNo ratings yet
- Predicting Clinical Outcomes From Genomics Deep Learning ModelsDocument11 pagesPredicting Clinical Outcomes From Genomics Deep Learning ModelsVenkat KunaNo ratings yet
- Rapid Dna Origami Nanostructure Detection and Classification Using The Yolov5 Deep Convolutional Neural NetworkDocument13 pagesRapid Dna Origami Nanostructure Detection and Classification Using The Yolov5 Deep Convolutional Neural NetworkEd swertNo ratings yet
- Analysis of Heart Disease Using in Data Mining Tools Orange and WekaDocument7 pagesAnalysis of Heart Disease Using in Data Mining Tools Orange and WekabekNo ratings yet
- Breast Cancer Detection Using SVM Classifier With Grid Search TechniqueDocument6 pagesBreast Cancer Detection Using SVM Classifier With Grid Search TechniqueroniwahyuNo ratings yet
- CNN-Based Image Analysis For Malaria Diagnosis: Abstract - Malaria Is A Major Global Health Threat. TheDocument4 pagesCNN-Based Image Analysis For Malaria Diagnosis: Abstract - Malaria Is A Major Global Health Threat. TheMiftahul RakaNo ratings yet
- Project (Sec: 01)Document10 pagesProject (Sec: 01)Amanat Khan ShishirNo ratings yet
- DATA MINING and MACHINE LEARNING. CLASSIFICATION PREDICTIVE TECHNIQUES: NAIVE BAYES, NEAREST NEIGHBORS and NEURAL NETWORKS: Examples with MATLABFrom EverandDATA MINING and MACHINE LEARNING. CLASSIFICATION PREDICTIVE TECHNIQUES: NAIVE BAYES, NEAREST NEIGHBORS and NEURAL NETWORKS: Examples with MATLABNo ratings yet
- Innovative Mathematical Insights through Artificial Intelligence (AI): Analysing Ramanujan Series and the Relationship between e and π\piDocument8 pagesInnovative Mathematical Insights through Artificial Intelligence (AI): Analysing Ramanujan Series and the Relationship between e and π\piInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Intrusion Detection and Prevention Systems for Ad-Hoc NetworksDocument8 pagesIntrusion Detection and Prevention Systems for Ad-Hoc NetworksInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- AI Robots in Various SectorDocument3 pagesAI Robots in Various SectorInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- An Explanatory Sequential Study of Public Elementary School Teachers on Deped Computerization Program (DCP)Document7 pagesAn Explanatory Sequential Study of Public Elementary School Teachers on Deped Computerization Program (DCP)International Journal of Innovative Science and Research TechnologyNo ratings yet
- Nurturing Corporate Employee Emotional Wellbeing, Time Management and the Influence on FamilyDocument8 pagesNurturing Corporate Employee Emotional Wellbeing, Time Management and the Influence on FamilyInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Fuzzy based Tie-Line and LFC of a Two-Area Interconnected SystemDocument6 pagesFuzzy based Tie-Line and LFC of a Two-Area Interconnected SystemInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Dynamic Analysis of High-Rise Buildings for Various Irregularities with and without Floating ColumnDocument3 pagesDynamic Analysis of High-Rise Buildings for Various Irregularities with and without Floating ColumnInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Importance of Early Intervention of Traumatic Cataract in ChildrenDocument5 pagesImportance of Early Intervention of Traumatic Cataract in ChildrenInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Geotechnical Assessment of Selected Lateritic Soils in Southwest Nigeria for Road Construction and Development of Artificial Neural Network Mathematical Based Model for Prediction of the California Bearing RatioDocument10 pagesGeotechnical Assessment of Selected Lateritic Soils in Southwest Nigeria for Road Construction and Development of Artificial Neural Network Mathematical Based Model for Prediction of the California Bearing RatioInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- The Effects of Liquid Density and Impeller Size with Volute Clearance on the Performance of Radial Blade Centrifugal Pumps: An Experimental ApproachDocument16 pagesThe Effects of Liquid Density and Impeller Size with Volute Clearance on the Performance of Radial Blade Centrifugal Pumps: An Experimental ApproachInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Integration of Information Communication Technology, Strategic Leadership and Academic Performance in Universities in North Kivu, Democratic Republic of CongoDocument7 pagesIntegration of Information Communication Technology, Strategic Leadership and Academic Performance in Universities in North Kivu, Democratic Republic of CongoInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- A Novel Approach to Template Filling with Automatic Speech Recognition for Healthcare ProfessionalsDocument6 pagesA Novel Approach to Template Filling with Automatic Speech Recognition for Healthcare ProfessionalsInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Breaking Down Barriers To Inclusion: Stories of Grade Four Teachers in Maintream ClassroomsDocument14 pagesBreaking Down Barriers To Inclusion: Stories of Grade Four Teachers in Maintream ClassroomsInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- The Influence of Artificial Intelligence On Employment Trends in The United States (US)Document5 pagesThe Influence of Artificial Intelligence On Employment Trends in The United States (US)International Journal of Innovative Science and Research TechnologyNo ratings yet
- Diode Laser Therapy for Drug Induced Gingival Enlaregement: A CaseReportDocument4 pagesDiode Laser Therapy for Drug Induced Gingival Enlaregement: A CaseReportInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Comparison of JavaScript Frontend Frameworks - Angular, React, and VueDocument8 pagesComparison of JavaScript Frontend Frameworks - Angular, React, and VueInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Analysis of Factors Obstacling Construction Work in The Tojo Una-Una Islands RegionDocument9 pagesAnalysis of Factors Obstacling Construction Work in The Tojo Una-Una Islands RegionInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Occupational Safety: PPE Use and Hazard Experiences Among Welders in Valencia City, BukidnonDocument11 pagesOccupational Safety: PPE Use and Hazard Experiences Among Welders in Valencia City, BukidnonInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Organizational Factors That Influence Information Security in Smes: A Case Study of Mogadishu, SomaliaDocument10 pagesOrganizational Factors That Influence Information Security in Smes: A Case Study of Mogadishu, SomaliaInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Disseminating The Real-World Importance of Conjunct Studies of Acculturation, Transculturation, and Deculturation Processes: Why This Can Be A Useful Technique To Analyze Real-World ObservationsDocument15 pagesDisseminating The Real-World Importance of Conjunct Studies of Acculturation, Transculturation, and Deculturation Processes: Why This Can Be A Useful Technique To Analyze Real-World ObservationsInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Mediating Effect of Encouraging Attitude of School Principals On Personal Well-Being and Career Ethics of TeachersDocument10 pagesMediating Effect of Encouraging Attitude of School Principals On Personal Well-Being and Career Ethics of TeachersInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- The Relationship of Total Quality Management Practices and Project Performance With Risk Management As Mediator: A Study of East Coast Rail Link Project in MalaysiaDocument19 pagesThe Relationship of Total Quality Management Practices and Project Performance With Risk Management As Mediator: A Study of East Coast Rail Link Project in MalaysiaInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Classifying Crop Leaf Diseases Using Different Deep Learning Models With Transfer LearningDocument8 pagesClassifying Crop Leaf Diseases Using Different Deep Learning Models With Transfer LearningInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- The Relationship Between Knowledge About Halitosis and Dental and Oral Hygiene in Patients With Fixed Orthodontics at The HK Medical Center MakassarDocument4 pagesThe Relationship Between Knowledge About Halitosis and Dental and Oral Hygiene in Patients With Fixed Orthodontics at The HK Medical Center MakassarInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Pecha Kucha Presentation in The University English Classes: Advantages and DisadvantagesDocument4 pagesPecha Kucha Presentation in The University English Classes: Advantages and DisadvantagesInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Total Cost of Ownership of Electric Car and Internal Combustion Engine Car With Performance NormalizationDocument11 pagesTotal Cost of Ownership of Electric Car and Internal Combustion Engine Car With Performance NormalizationInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Integrating Multimodal Deep Learning For Enhanced News Sentiment Analysis and Market Movement ForecastingDocument8 pagesIntegrating Multimodal Deep Learning For Enhanced News Sentiment Analysis and Market Movement ForecastingInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- An Investigation Model To Combat Computer Gaming Delinquency in South Africa, Server-Based Gaming, and Illegal Online GamingDocument15 pagesAn Investigation Model To Combat Computer Gaming Delinquency in South Africa, Server-Based Gaming, and Illegal Online GamingInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Gender-Based Violence: Engaging Children in The SolutionDocument10 pagesGender-Based Violence: Engaging Children in The SolutionInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Development of Creative Thinking: Case Study of Basic Design StudioDocument9 pagesDevelopment of Creative Thinking: Case Study of Basic Design StudioInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- PDF Deep Reinforcement Learning in Action 1St Edition Alexander Zai Ebook Full ChapterDocument53 pagesPDF Deep Reinforcement Learning in Action 1St Edition Alexander Zai Ebook Full Chapterrick.kelleher522100% (3)
- Multimae: Multi-Modal Multi-Task Masked AutoencodersDocument21 pagesMultimae: Multi-Modal Multi-Task Masked AutoencodersAnya PankovaNo ratings yet
- Artificial Intelligence in Food Industry: Submitted To: Sir Anwar Submitted By: Gul E Kainat 15-ARID-4939Document32 pagesArtificial Intelligence in Food Industry: Submitted To: Sir Anwar Submitted By: Gul E Kainat 15-ARID-4939Lubna KhanNo ratings yet
- How To Use Google Colaboratory For Video Processing - CodeProjectDocument12 pagesHow To Use Google Colaboratory For Video Processing - CodeProjectgfgomesNo ratings yet
- Play With Data ScienceDocument306 pagesPlay With Data SciencedpsharmaNo ratings yet
- ML - Expectation-Maximization AlgorithmDocument3 pagesML - Expectation-Maximization AlgorithmprakashNo ratings yet
- CART v6.0Document434 pagesCART v6.0segorin2No ratings yet
- Artificial Intelligence Machine Learning Program BrochureDocument24 pagesArtificial Intelligence Machine Learning Program BrochureSunnyNo ratings yet
- Deep Learning For Prediction of Depressive Symptoms in A Large Textual DatasetDocument24 pagesDeep Learning For Prediction of Depressive Symptoms in A Large Textual DatasetAmith PrasanthNo ratings yet
- Optimization in Machine LearningDocument26 pagesOptimization in Machine LearningrbernalMxNo ratings yet
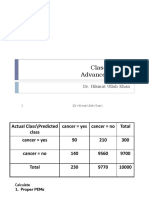
- 07 ML Classificaion Advanced KappaDocument18 pages07 ML Classificaion Advanced KappaIn TechNo ratings yet
- B.tech 20-21 InternshipDocument9 pagesB.tech 20-21 Internship20501A05D5 NASIKA PRAVALLIKANo ratings yet
- Keystroke DynamicsDocument6 pagesKeystroke DynamicsrasbiakNo ratings yet
- Dissertation Research Aims and ObjectivesDocument7 pagesDissertation Research Aims and ObjectivesHelpWritingACollegePaperCanada100% (1)
- Book Review:: Pedro Domingos. Basic Books. 2015. ISBN 978-0465065707Document2 pagesBook Review:: Pedro Domingos. Basic Books. 2015. ISBN 978-0465065707Anurag GuptaNo ratings yet
- A Paper On Stylometric Differences Between Authentic and Fabricated AhadithDocument18 pagesA Paper On Stylometric Differences Between Authentic and Fabricated AhadithMuhammad Ammar adilNo ratings yet
- Modeling PROTAC Degradation Activity With Machine LearningDocument12 pagesModeling PROTAC Degradation Activity With Machine Learningpradeep kumarNo ratings yet
- Written Work 1 - Attempt Review3Document6 pagesWritten Work 1 - Attempt Review3RussianOmeleteNo ratings yet
- Machine Learning Made Easy: A Review of Scikit-Learn Package in Python Programming LanguageDocument14 pagesMachine Learning Made Easy: A Review of Scikit-Learn Package in Python Programming Languagejuan carlosNo ratings yet
- Deep Learning - Lesson PlanDocument5 pagesDeep Learning - Lesson PlanAnonymous xMYE0TiNBcNo ratings yet
- Machine Learning Algorithms For Signal and Image Processing Suman Lata Tripathi Full ChapterDocument67 pagesMachine Learning Algorithms For Signal and Image Processing Suman Lata Tripathi Full Chapterdolores.godfrey250100% (7)
- Machine Learning PaperDocument5 pagesMachine Learning PaperAshutoshNo ratings yet
- 2022 - Predicting Tunnel Squeezing Using Support Vector Machine Optimized by Whale Optimization AlgorithmDocument24 pages2022 - Predicting Tunnel Squeezing Using Support Vector Machine Optimized by Whale Optimization Algorithm周牮No ratings yet
- Prediction of Reservoir Brittleness From Geophysical Logs Using Machine Learning AlgorithmsDocument16 pagesPrediction of Reservoir Brittleness From Geophysical Logs Using Machine Learning Algorithmsdobeng147No ratings yet
- Deep Face Recognition: A Survey: Mei Wang, Weihong DengDocument31 pagesDeep Face Recognition: A Survey: Mei Wang, Weihong DengGitaNo ratings yet
- ThoughtSpot SpotIQ AI Driven Analytics White Paper PDFDocument20 pagesThoughtSpot SpotIQ AI Driven Analytics White Paper PDFfoliox100% (1)
- Lecture 1 - Introduction To MLDocument25 pagesLecture 1 - Introduction To MLsba20050No ratings yet
- Machine Learning Using Python - Discover The World of MLDocument108 pagesMachine Learning Using Python - Discover The World of MLbahiniy286No ratings yet




















































































































Volume 8, Issue 12, December 2023 International Journal of Innovative Science and Research Technology
ISSN No:-2456-2165
Optimized Gene Classification using Support
Vector Machine with Convolutional
Neural Network for Cancer Detection
from Gene Expression Microarray Data
Vishwas Victor; Dr. Ragini Shukla
Department of IT & CS., Dr. C. V. Raman University Kota, Bilaspur, India
Abstract:- There are numerous approaches for the dataset's dimensionality, [13]. Microarray gene
handling microarray gene expression data since new expression studies frequently produce a large number of
feature selection techniques are constantly being characteristics for a limited number of patients, resulting in
developed. To create a new subset of pertinent features, a high dimensional dataset with a small number of samples.
feature selection (FS) is utilized to pinpoint the essential Gene expression data is extremely complicated and
feature subset. The model that used the informative problematic; genes are connected with one other either
subset projected that a classification model generated directly or indirectly, making the classification process
solely using this subset would have higher predicted extremely tough and challenging. Typically, this means
accuracy than a model developed using the whole employing a precise and potent feature selection technique
collection of attributes. is required [10].
We offer an analytical approach for cancer The field of Deep Learning (DL) is concerned with
classification and developed a model using Support using deep networks for information processing.
Vector Machine as classifier and after that Convolutional Neural Networks (CNNs) are a type of
Convolutional Neural Network in the aspect of Deep artificial neural network that can extract local features from
Learning. The outcome received in the context of the data. CNN assigns weights based on a single feature
proposed model is very impressive and accurate. mapping, simplifying the network model and allowing for a
reduction in overall weights. DL is designed to handle data
Keywords:- Feature selection; Optimization; Classification; using both supervised and unsupervised methods, with
Support Vector Machine (SVM); Deep Learning; Machine learning taking place on several layers of features and
Learning; Convolutional Neural Network (CNN). descriptions [17]. The processes of feature extraction and
selection result in a distilled set of the essential
I. INTRODUCTION characteristics that define the core characteristics of the
This A microarray expression experiment records the data and categorize it. You can carry out this classification
expression levels of thousands of genes simultaneously; with or without supervision. While supervised learning uses
each gene is a segment of DNA that carries all the data with output class markers, unsupervised learning uses
information needed to make several kinds of proteins in our information without output class labels. An algorithm
body. The main methods used in these experiments are illustrates the relationship between patterns in the input
explained by authors that either multiple monitoring of each attribute variables and the associated descriptors in the
gene under various conditions, or evaluating each gene in a output for supervised approaches [5]
single environment but in different types of tissues, The main objective of this literature to get better
particularly cancerous tissues [9]. One method for reducing results with highest accuracy for feature selection of gene
classifier calculation errors is feature selection (FS), which expression microarray data. Lots of works happened till
removes noisy, redundant, and unrelated qualities from the date, but at this end for the humanity it is required to re-
original data set and selects important attributes. According examine the datasets with better prediction related to
to the authors, generally feature selection techniques fall identify the cancer form the infected gene expression
into three main categories: wrapper, filter, and embedded microarray data. Here, we have proposed an embedded
models [7]. approach for Dimension Reduction and Classification using
The microarray dataset technique faces two primary Support Vector Machine with Convolutional Neural
issues: an excessive number of genes relative to a lesser Network (DRC-SVM-CNN). This approach has produced
number of samples. The process of identifying relevant the result with better accuracy and prediction.
features from the data and displaying the higher dimension This literature contains the following sections: In
dataset with a condensed search space is known as feature Section 2, the Microarray Technology and Datasets
selection (FS). The proper FS resolution for microarray described. In Section 3, Related works compiled by the
data, however, is very difficult to determine because the researchers and their published articles are evaluated. In
sample size is smaller than the total number of genes. There Section 4 Proposed Methods and model which are used to
are a number of things to take into account when reducing address the challenge of features selection, explained. In
IJISRT23DEC1319 www.ijisrt.com 1375
Volume 8, Issue 12, December 2023 International Journal of Innovative Science and Research Technology
ISSN No:-2456-2165
Section 5, implementation and analysis explained in this unknown individuals—normal and abnormal—must be
section. Section 6 consists result analysis and discussion, obtained. These are cautiously placed into the silicon chip's
obtained through proposed model. In Section 7, conclusion little openings. Following that, DNAs hybridize. Annealing
of the literature explained. primer is used to separate the RNA, mRNA, and tRNA
from these DNAs, which are linked to their complementary
II. MICROARRAY TECHNOLOGY AND DNA. A tagged cDNA is produced following the
DATASETS transcriptase enzyme's labeling of mRNA. After the tagged
cDNA hybridizes, a green or red laser light is seen on the
It is intended to apply a tissue or DNA sample to the chip. Eventually, the intensity is calculated and microarray
chip's minuscule areas. Deoxyribonucleic acid, or DNA, is data is produced.
a type of genetic material found in nearly all living things,
including humans. A throughput technique used in cancer In the given figure 1, the process to create gene
research for illness detection and prognosis is microarray expression microarray data is illustrated. First, we collect
data. Microarray technology is the most straightforward the sample from the infected person, then by applying them
method for identifying patterns in both normal and aberrant through microarray silicon chip, we got the microarray
tissue. A silicon device called a microarray displays data.
thousands of small dots. First, several genetic samples from
Fig. 1: Gene expression microarray data creation process
These are the address of gene expression microarray https://web.stanford.edu/~hastie/CASI_files/DATA/leu
datasets repository from where we can get them for our kemia.html
experiments- https://jundongl.github.io/scikit-feature/datasets.html,
https://www.ncbi.nlm.nih.gov https://vizgen.com/data-release-program/
http://csse.szu.edu.cn/staff/zhuzx/Datasets.html https://dataverse.harvard.edu/
http://www.biolab.si/supp/bi-cancer/projections/
Our analysis has been done on these datasets given in
the Table I.
Table 1: Description of Microarray Datasets
Dataset Features Sample No. of Classes
Breast 24,481 78 2
Prostate 12,600 102 2
Brain 12,625 21 2
CNS 7129 60 2
Colon 2000 62 2
DLBCL 4026 47 2
GLI 22,283 85 2
Ovarian 15,154 253 2
Leukemia 7070 72 2
IJISRT23DEC1319 www.ijisrt.com 1376
Volume 8, Issue 12, December 2023 International Journal of Innovative Science and Research Technology
ISSN No:-2456-2165
III. RELATED WORKS The authors have proposed a model using statistical
methods and machine learning for feature selection.
For choosing a set of microarray data, a minimum Support Vector Machine (SVM) was the one method for
collection of features or genes (or subsets of features), a selection of the features subset [5]. Finding this genetic
variety of strategies have been put forth by various material from the entire genome is essential, and depending
scholars. After conducting a thorough literature review, we on how these few genes express themselves, a patient's
will have a brief discussion regarding current methods as condition can be classified as either having or not. Author
they pertain to our suggested work. highlighted the various challenges associated with the gene
expression data in the presence of noise and missing values.
Authors have proposed an algorithm based on Deep
Neural Network using Convolutional Neural Network[1]. Authors have explained a deep feedforward approach
They implemented the technique to recover overfitting for categorizing provided microarray cancer data into a set
problem in DNN. Authors explained the complexity of the of classifications for eventual diagnosis [6]. Authors
gene expression microarray datasets, as these datasets employed a 7-layer deep neural network design with
consists huge number of features. They described about different parameters on all datasets. The difficulties of very
their proposed improved-Deep Neural Network algorithm less sample size and high dimensionality have been solved
to find out the relevant features to predict the best solutions. by using a popular dimension reduction method
named principal component analysis. The Min-Max
Authors have described the nature of gene expression methodology is used to scale the feature values, and the
microarray datasets, which is contains lots of information suggested method is verified on eight common microarray
under it [2]. Gene expression microarray datasets are high cancer datasets. A comparison with state-of-the-art
dimensional datasets and it is challenging to get the most approaches is conducted, and the effectiveness of the
relevant features predict the probabilities of disease. suggested methodology outperforms several of the existing
Authors have proposed Deep Gene Selection (DGS) methods.
algorithm for the best performance to achieved
classification of cancer. They compared various gene Authors have explained some of the most recent
selection methods with DGS and got the best results from feature selection approaches for analyzing the microarray
DGS with better accuracy and computation cost. datasets to produce better results [7]. Author described that
the large number of features and limited quantity of
Utilizing sophisticated classification and prediction samples, microarray data classification presents a
methods, microarray datasets, which contain information significant task for machine learning researchers. Author
about the various gene expression patterns have been offered an experimental assessment on the most significant
studied, in order to help with the crucial and difficult task datasets utilizing recognized feature selection methods,
of early diagnosis of chronic diseases like cancer. To with the goal of facilitating comparative investigation by
address the high-dimensional microarray datasets problem the research society rather than providing the optimal
and ultimately improve the accuracy of cancer feature selection approach.
classification, a hybrid filter-genetic feature selection
strategy has been presented by the authors [3]. To further Authors have defined that comparison of the period
optimize and enrich the chosen features and increase the of evaluation, classification accuracy, and possibilities to
suggested method's capacity for cancer classification, a recognize the disease, as well as calculating the strictness of
genetic algorithm has been used. According to the the positions of disease, the experimental outputs show that
experimental findings, the suggested hybrid filter-genetic deep neural network classification execution can be better
feature selection strategy outperformed a number of widely than present classification techniques [8]. The authors have
used machine learning techniques to obtain accuracy, explained about the necessary to take a close look at this
recall, F-measure, and precision. topic, as well as the research findings and related issues, in
order to achieve a perspective of the ailment categories.
Gene expression profiling, or microarrays, evaluate The author have described, Neural networks are the
and identify patterns and levels of gene expression in a foundation of machine learning, which includes deep
variety of cell types and tissues. Author explained the learning. Knowledge, or the ability to distinguish between
Cancer classification using DNA microarray technology, different types of information and organize it into a manner
which allows for the simultaneous intense treatment of that is easily comprehended by humans, has been retrieved
hundreds of gene expressions on a single chip. The Latent from the massive amount of raw data. Cancer prediction is
Feature Selection Technique is proposed in the suggested deep learning's primary function.
method by the author, to shorten classification times and
improve accuracy after deep learning algorithms are trained Authors have proposed a method on the basis of
on microarray data to extract features [4]. This study used Support Vector Machine (SVM) and Mutual Information
bone marrow PC gene expression data to develop the (MI). The authors explained that SVM is a technique for
Artificial Bee Colony (ABC) feature selection method. supervised learning that can handle challenging
Author found the tumor classification, performed using classification issues, whereas the informative genes can be
Convolutional Neural Networks has a better accuracy. determined using the mutual information (MI) among the
genes and the class label. By selecting the most useful gene
subset, the suggested method improves classification
IJISRT23DEC1319 www.ijisrt.com 1377
Volume 8, Issue 12, December 2023 International Journal of Innovative Science and Research Technology
ISSN No:-2456-2165
accuracy while reducing the dimension of the input cancer research as well as illness prognosis and treatment
characteristics when compared to alternative methods [9]. [16]. The authors have described, Neural networks serve as
the foundation for deep learning, which is a kind of
Authors have developed A hybrid cancer automated learning. The data has been extracted from the
classification method by using number of machine learning extensive body of knowledge that is raw data, allowing for
techniques, including the easy-to-understand Decision Tree discrimination and easy comprehension within a
classifier that doesn't need a parameter, Grid Search CV framework.
(cross-validation) to optimize the maximum depth
hyperparameter, and Pearson's correlation coefficient as a Authors have developed a model named hybrid
correlation-based feature selection and reducer. To assess mSVM-RFE-iRF for gene selection and classification using
the approach, seven common microarray cancer datasets are Support Vector Machine (SVM), Recursive Feature
employed. The results show that the suggested technique Elimination (RFE) and improved Random Forest (iRF) and
chooses the most informative characteristics, boosts compared with Convolutional Neural Network (CNN).
classification accuracy, and significantly reduces the Deep learning algorithm applications are receiving a lot of
number of genes needed for classification [9]. interest as a means of resolving various AI-related
problems. According to author, when compared to hybrid
Authors have introduced an efficient feature selection mSVM-RFE-iRF, the majority of experimental results on
technique that explores the nonlinear mapping skills of cancer datasets showed that CNN is more accurate and
double RBF-kernels in conjunction with weighted analysis minimizes gene when it comes to cancer classification [17].
for obtaining feature genes from gene expression data [11].
Outperformed earlier approaches with comparatively higher Our goal is to provide an effective model that can
accuracy, true positive rate, false positive rate, and less choose the smallest feature set with the highest degree of
runtime, proposed method tested on four benchmark accuracy. To that end, we have taken into consideration the
datasets with either two-class phenotypes or multiclass deep learning technique known as Convolutional Neural
phenotypes. The authors explained, with a large number of Network, executed on optimized genes collected by
genes and limited samples, the main difficulty in analyzing Support Vector Machine and Random Forest classifiers.
gene expression data is separating disease-related
information from a vast amount of noise and redundant IV. PROPOSED TECHNIQUES
data. To solve this issue, removing unnecessary and
duplicate genes by gene selection has been a crucial step. Finding the w and b parameters that define the
hyperplane in a way that optimizes the margin which is the
Authors have suggested a method to significantly distance between the hyperplane and the closest data points
reduce the data source's dimension in order to increase the from each class is the primary goal. Support vectors are
accuracy of decreasing dimensionality. The study classifies defined as the set of these nearest neighbors. Followings are
feature selection algorithms into three categories: semi- the steps taken by us for analysis of the gene expression
supervised, supervised, and un-supervised learning. The data:
performance of multiple described methods in the literature
was analyzed, as well as the recent efforts to reduce the A. Dataset partitioning:
features for tumor diagnosis [12]. Three subsets of the dataset were created: a first set of
features for training, a second set for validation, and a third
Limited instances with a significant amount of set for testing. While the training dataset (first one) is used
imbalance between the classes can occasionally be found in to train the classifier that is utilized (algorithm), the
gene expression data. This may restrict a classification validation dataset is used to evaluate the performance of the
model's exposure to examples from other categories, which classifier and is applied in the optimization efficiency. In
may have an impact on the model's performance, explained order to assess the effectiveness of the entire feature
by the authors. Authors have developed an algorithm for selection and classifier procedure, the test dataset (final
feature selection by employing numerous iterations of 5- dataset) is employed.
fold cross-validation on various microarray datasets. The
algorithms' performance varied according to the data and B. Decreasing dimensionality of datasets:
feature reduction method applied [14]. Authors suggested a By using measurement approaches to find features that
method to identify cancer by using data were essential in the initial dataset for categorizing a target,
preprocessing methods such feature selection, the dataset is reduced.
oversampling, and classification models. The six datasets C. Assigning values for specifying parameters:
that were evaluated in the study were oversampled using The feature selection approach creates meaningful
SVMSMOTE. feature subsets by selecting important characteristics from
The authors have explained one of the best tools for the original dataset after removing unnecessary, redundant,
addressing the identification of cancer, prediction, and and noisy features. The potential feature subsets are
treatment is microarray gene-based expression profiling evaluated to find out classification accuracy. An
techniques. In healthcare, microarray research is used for explanation of the study technique is provided in Figure 2:
IJISRT23DEC1319 www.ijisrt.com 1378
Volume 8, Issue 12, December 2023 International Journal of Innovative Science and Research Technology
ISSN No:-2456-2165
Fig. 2: Proposed (DRC-SVM-CNN) algorithm flow diagram for classification
The conclusion will consequently be minimum feature b is the bias term that shifts the hyperplane away from
size and highest classification accuracy in order to acquire the origin.
the best feature subsets (standard deviation and average
number of features). Finding the values of w and b that maximize the
margin, the distance between the hyperplane and the closest
D. Applied Methodologies data points from each class, is the aim of defining the
Here we are explaining the applied methodologies for hyperplane. Support vectors are those closest data points.
the analysis of the research.
The following is a formulation for the SVM
Support Vector Machine optimization:
The foundation of Support Vector Machines (SVMs) is
a mathematical formulation that looks for the best Yi (w ⋅ xi + b) ≥ M, for i = 1, 2, …, N (2)
hyperplane to divide two data classes. An SVM's principal
mathematical expression is as follows: Where:
M is the margin (The distance apart the two parallel
Given a dataset with input data points xi and their hyperplanes are).
corresponding labels yi ∈ {−1,1} w and b are the parameters to be optimized.
where i =1, 2, …, N, an SVM seeks to find a Convolutional Neural Network
hyperplane defined by: Convolutional neural networks, or CNNs, are a kind of
deep learning models that are mainly utilized for tasks
w⋅x + b = 0 (1) involving images, however they can also be used with other
w is the weight vector that is orthogonal to the kinds of data. Figure 4 illustrating the typical architecture
hyperplane. of Convolutional Neural Network (CNN).
x represents the input features of a data point.
Fig. 3: Convolutional Neural Network Architecture
The following are the essential elements of a typical The mathematical representation of the convolution
CNN's mathematical expression: operation for a 2D input, such as an image, is as follows:
Convolution Operation (I * K) (i, j) =∑m ∑n I (i + m, j + n). K (m, n)
Convolution, the primary function of a CNN, is the (3)
process of extracting local characteristics from input data
by swiping a filter, sometimes referred to as a kernel, across
the data.
IJISRT23DEC1319 www.ijisrt.com 1379
Volume 8, Issue 12, December 2023 International Journal of Innovative Science and Research Technology
ISSN No:-2456-2165
Where: Depending on the task at hand, a loss function such as
I input. cross-entropy for classification or mean squared error
K is the convolutional kernel. (MSE) for regression should be used.
(i,j) represents the pixel location in the output feature
map. Back propagation and Optimization
(m,n) represents the pixel location in the kernel. CNNs are trained by varying the model's parameters
(weights and biases) to minimize the loss function through
Activation Function the use of optimization algorithms such as stochastic
After convolution, an activation function (commonly gradient descent and backpropagation.
ReLU - Rectified Linear Unit) is applied element-wise to
introduce non-linearity into the model. Mathematically, this Proposed Algorithm (DRC-SVM-CNN)
can be represented as: Collect Gene Expression Microarray Datasets
Preprocess the datasets for analysis
ReLU(x) = max (0, x) (4) Apply Support Vector Machine for classification
For optimization, select dropouts to reduce the
Pooling Operation overfitting problems
Pooling (e.g., max-pooling or average-pooling) is used Apply and tarin the autoencoder to reduce feature
to down-sample the feature maps, reducing the spatial dimension
dimensions and the number of parameters. Again, train SVM classifier on the reduced features and
analyse
Mathematically, max-pooling can be represented as:
Create a Convolutional neural network model for taking
Max Pooling(x) = max (neighbors of x) (5) reduced features as input Utilizing the encoded features,
train the convolutional neural network to perform
Fully Connected Layers classification without overfitting.
After several convolutional and pooling layers, CNNs
often have one or more fully connected layers to make V. IMPLEMENTATION AND ANALYSIS
high-level predictions.
To get experimental results, we have implemented our
A fully connected layer can be expressed proposed method using Python 3.9 version and Jupyter
mathematically as a matrix multiplication followed by an Notebook. We have used computer system with Intel i5,
activation function, typically a softmax for classification 1.80 GHz processor, 16GB RAM and Windows 11
tasks. environment. The outcomes achieved by the experimental
results are provided here as we have collected.
Loss Function
The error separating the genuine values from the Let us consider the following:
predicted values is measured by the loss function.
Fig. 4: Observing the Test and Validation loss over the Epochs
IJISRT23DEC1319 www.ijisrt.com 1380
Volume 8, Issue 12, December 2023 International Journal of Innovative Science and Research Technology
ISSN No:-2456-2165
Figure 4 represents the training and testing loss that the training loss is also very less and negligible, so that
evaluation, where we have found that validation loss is the training of model is in appropriate condition. Without
approximately at 0 constantly, whereas training loss is implementation of dropout selection and SVM, it found
fluctuating between 0.2 and -0.2 in each epochs due to the very uncertain.
high dimensional data cause overfitting. Here we can say
Fig. 5: Result Accuracy Representation of Data
Figure 5 represents the accuracy over all epochs and Specificity = (True Negatives) / (True Negatives +
we observed that the validation accuracy is 100% and False Positives)
training accuracy is at nearly 97%. So, that in this graphical
representation our proposed model has get fulfilled the Specificity is important when you want to minimize
expectations. In this regards we have analysed that the false positives.
proposed model is outperformed, one of the better solution
for optimization and classification of gene expression C. Accuracy
microarray data. Accuracy is a measure of overall correctness. It
calculates the proportion of all cases (both positive and
VI. RESULTS ANALYSIS AND DISCUSSION negative) that the model classifies correctly. It is calculated
as:
According to the proposed model the classification
accuracy of original feature sets are very impressive. Accuracy = (True Positives + True Negatives) / (True
Tensorflow, Numpy, Scikit-Learn are used in our proposed Positives + True Negatives + False Positives + False
program. To achieve these results, datasets were divided for Negatives)
experimental purpose into two parts that are Training and
Testing, such as 80:20 ratios. Sensitivity, Specificity, and Accuracy provides a broad view of a model's
Accuracy are checked for performance evaluation. performance but may not be the best metric when class
imbalances exist or when certain types of errors are more
A. Sensitivity costly than others.
Sensitivity measures the proportion of actual positive
cases that the model correctly identifies as positive. It is D. Results from Selected Datasets
calculated as: The following results have described the experimental
findings for each microarray dataset. After optimization of
Sensitivity = (True Positives) / (True Positives + False gene expression microarray data by selecting dropouts and
Negatives) applying Support Vector Machine with Convolutional
Neural Network, our model gets trained and analysed by
Sensitivity is important when you want to minimize calculating three tests for Sensitivity, Specificity, and
false negatives, as in medical diagnoses, where missing a Accuracy. Here we can see the collected results with
true positive (e.g., a disease) could be critical. classification accuracy almost above 99% on average. So,
that the performance of the proposed model is very much
B. Specificity suitable for the prediction of the disease identification
Specificity measures the proportion of actual negative through gene expression microarray data. The performance
cases that the model correctly identifies as negative. It is assessments of the complete microarray dataset are
calculated as: mentioned in Table 2.
IJISRT23DEC1319 www.ijisrt.com 1381
Volume 8, Issue 12, December 2023 International Journal of Innovative Science and Research Technology
ISSN No:-2456-2165
Table 2: Datasets used in the experimental study and obtained results
Datasets Genes Selected Sensitivity Specificity Accuracy
Breast 4,896 97.68 97.86 99.79
Prostate 2,520 97.12 98.35 98.55
Brain 2,525 98.03 98.41 99.03
CNS 1425 97.62 98.89 99.15
Colon 400 98.93 98.3 100
DLBCL 805 97.62 97.79 98.64
GLI 4,456 96.64 98.73 99.62
Ovarian 3,031 97.68 98.2 99.74
Leukaemia 1414 98.68 98.81 99.91
VII. CONCLUSION [5]. S. Arora, and S. Gupta, “Feature Selection of Gene
Expression Data Using Machine Learning and
In recent years, there has been an increase in the Statistical Modelling,” International Journal of Recent
occurrence of challenging diseases, the significance of Scientific Research, Vol. 12, Issue, 10 (B), pp. 43334-
producing microarray data from different tissues, and the 43340, 2021.
analysis of this data. As we know the challenge of [6]. H. S. Basavegowda, and G. Dagnew, “Deep Learning
uncertainty of the microarray gene expression data due to Approach for Microarray Cancer Data Classification,”
huge number of features and very small number of CAAI Transactions on Intelligence Technology, vol.
instances. To overcome this problem, it is required to 5(1), pp. 22–33, 2020.
optimize the data and reduce the irrelevant and redundant [7]. V. Bolón-Canedo, N. Sánchez-Maroño, A. Alonso-
data from datasets, having huge number of features or Betanzos, J. M. Benítez, & F. Herrera, “A Review of
dimension. We have worked in this research, to optimize Microarray Datasets and Applied Feature Selection
the gene expression microarray data using our proposed Methods,” Information Sciences, vol. 282, pp. 111–
method by implementation of SVM and dropout selection, 135. 2014.
thereafter obtained optimized data has processed through [8]. V. Chandrasekar, V. Sureshkumar, T. S. Kumar, and
convolutional neural network. This proposed model is S. Shanmugapriya, “Disease Prediction Based on
named by us as DRC-SVM-CNN can be identified as Micro Array Classification using Deep Learning
embedded approach for dimension reduction and Techniques,” Microprocessors and Microsystems, 77,
classification using support vector machine (SVM) with 2020.
convolutional neural network (CNN). The SVM classifier [9]. C. Devi Arockia Vanitha, D. Devaraj, and M.
provides enhanced results after creating a new feature set. Venkatesulu, “Gene Expression Data Classification
This result has been contrasted with classifier accuracy using Support Vector Machine and Mutual
beyond sum dataset. Selected genes from nine cancer Information-Based Gene Selection,” Procedia
datasets were to be chosen using a convolutional neural Computer Science, vol. 47(C), pp. 13–21. 2014.
network (CNN). Prior to classifying and choosing the right [10]. H. Fathi, H. Alsalman, A. Gumaei, I. I. M. Manhrawy,
genes in each dataset, it is imperative to pick informative A. G. Hussien, and P. El-Kafrawy, “An Efficient
genes accurately. Using our model, we have obtained on Cancer Classification Model using Microarray and
average 99% accuracy among all nine datasets. Certainly, High-Dimensional Data,” Computational Intelligence
we can say that, this model (DRC-SVM-CNN) has and Neuroscience, 2021.
produced all time better results in the area of gene [11]. S. Liu, C. Xu, Y. Zhang, J. Liu, B. Yu, X. Liu, and M.
expression microarray datasets for cancer classification. Dehmer, “Feature Selection of Gene Expression Data
for Cancer Classification using Double RBF-Kernels,”
REFERENCES BMC Bioinformatics, 19(1), 2018.
[1]. O. Ahmed and A. Brifcani, “Gene Expression [12]. N. Mahendran, P. M. Durai Raj Vincent, K.
Classification Based on Deep Learning,” 4th Srinivasan, and C. Y. Chang, “Machine Learning
Scientific International Conference Najaf, SICN, pp. Based Computational Gene Selection Models: A
145–149, 2019. Survey, Performance Evaluation, Open Issues, and
[2]. R. Alanni, J. Hou, H. Azzawi, and Y. Xiang. “Deep Future Research Directions,” Frontiers in Genetics,
Gene Selection Method to Select Genes from Vol. 11, 2020.
Microarray Datasets for Cancer Classification,” BMC [13]. Malibari, R. M. Alshehri, F. N. Al-Wesabi, N. Negm,
Bioinformatics, vol. 20(1), 2019. M. Al Duhayyim, A. M. Hilal, I. Yaseen, and A.
[3]. W. Ali, and F. Saeed, “Hybrid Filter and Genetic Motwakel, “Deep Learning Enabled Microarray Gene
Algorithm-Based Feature Selection for Improving Expression Classification for Data Science
Cancer Classification in High-Dimensional Applications,” Computers, Materials and Continua,
Microarray Data,” Processes, 11(2), 2023. vol. 73(2), pp. 4277–4290, 2022.
[4]. H. Z. Almarzouki, “Deep-Learning-Based Cancer [14]. O. O. Petinrin, F. Saeed, N. Salim, M. Toseef, Z. Liu,
Profiles Classification Using Gene Expression Data and I. O. Muyide, “Dimension Reduction and
Profile,” Journal of Healthcare Engineering, 2022. Classifier-Based Feature Selection for Oversampled
IJISRT23DEC1319 www.ijisrt.com 1382
Volume 8, Issue 12, December 2023 International Journal of Innovative Science and Research Technology
ISSN No:-2456-2165
Gene Expression Data and Cancer Classification,”
Processes, vol. 11(7), 2023.
[15]. S. H. Shah, M. J. Iqbal, I. Ahmad, S. Khan, and J. J. P.
C. Rodrigues, “Optimized Gene Selection and
Classification of Cancer from Microarray Gene
Expression Data using Deep Learning,” Neural
Computing and Applications, 2020.
[16]. Shyamala Gowri, S. Bhuvaneshwari, A. Abirami, R.
Dilli Rani, K. N. Anirudh, and S. Keerthi Shree,
“DNA Microarray for Cancer Classification Using
Deep Learning,” European Chemical Bulletin, pp.
1631–1641, 2023.
[17]. Q. Zeebaree, H. Haron, and A. M. Abdulazeez, “Gene
Selection and Classification of Microarray Data Using
Convolutional Neural Network,” International
Conference on Advanced Science and Engineering
(ICOASE), October 2018.
IJISRT23DEC1319 www.ijisrt.com 1383
You might also like
- Graham Giller Wilmott TalkDocument31 pagesGraham Giller Wilmott TalkGraham GillerNo ratings yet
- Cs 229, Autumn 2016 Problem Set #2: Naive Bayes, SVMS, and TheoryDocument20 pagesCs 229, Autumn 2016 Problem Set #2: Naive Bayes, SVMS, and TheoryZeeshan Ali SayyedNo ratings yet
- Gene Selection and Classification of Microarray Data Using Convolutional Neural NetworkDocument6 pagesGene Selection and Classification of Microarray Data Using Convolutional Neural NetworkTajbia HossainNo ratings yet
- Cancer Detection Using Convolutional Neural Network: February 2021Document10 pagesCancer Detection Using Convolutional Neural Network: February 2021worldworld312No ratings yet
- Almugren, Alshamlan - 2019 - A Survey On Hybrid Feature Selection Methods in Microarray Gene Expression Data For Cancer ClassificationDocument16 pagesAlmugren, Alshamlan - 2019 - A Survey On Hybrid Feature Selection Methods in Microarray Gene Expression Data For Cancer Classificationiyousafzai1No ratings yet
- Hybrid Model For Detection of Brain Tumor Using Convolution Neural NetworksDocument7 pagesHybrid Model For Detection of Brain Tumor Using Convolution Neural NetworksCSIT iaesprimeNo ratings yet
- A Microarray Gene Expression Data Classification UDocument14 pagesA Microarray Gene Expression Data Classification UHuseyin OztoprakNo ratings yet
- Enhanced Network Anomaly Detection Based On Deep Neural NetworksDocument16 pagesEnhanced Network Anomaly Detection Based On Deep Neural NetworksYishak TadeleNo ratings yet
- Brain Tumour Detection Using M-IRO-Journals-3 4 5Document12 pagesBrain Tumour Detection Using M-IRO-Journals-3 4 5MR. AJAYNo ratings yet
- Performance Analysis and Evaluation of Different Data Mining Algorithms Used For Cancer ClassificationDocument7 pagesPerformance Analysis and Evaluation of Different Data Mining Algorithms Used For Cancer ClassificationPAUL AKAMPURIRANo ratings yet
- Irjet V10i214Document3 pagesIrjet V10i214tanzimreadsNo ratings yet
- Astuti 2018 J. Phys. Conf. Ser. 971 012003Document8 pagesAstuti 2018 J. Phys. Conf. Ser. 971 012003fauzieahmad526No ratings yet
- EtextpathshaalaDocument21 pagesEtextpathshaalaMIGUELNo ratings yet
- 1 s2.0 S0079610722000803 MainDocument13 pages1 s2.0 S0079610722000803 MainsznistvanNo ratings yet
- CRISP-MED-DM A Methodology of Diagnosing Breast CancerDocument13 pagesCRISP-MED-DM A Methodology of Diagnosing Breast CancerInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Genes PaperDocument11 pagesGenes PaperTomas PetittiNo ratings yet
- Comparation Analysis of Ensemble Technique With Boosting (Xgboost) and Bagging (Randomforest) For Classify Splice Junction Dna Sequence CategoryDocument10 pagesComparation Analysis of Ensemble Technique With Boosting (Xgboost) and Bagging (Randomforest) For Classify Splice Junction Dna Sequence CategoryFatrinaNo ratings yet
- FsNET FEature Selection Network On HDDDocument14 pagesFsNET FEature Selection Network On HDDnfbeusebio3962No ratings yet
- Diagnosis of Chronic Brain Syndrome Using Deep LearningDocument10 pagesDiagnosis of Chronic Brain Syndrome Using Deep LearningIJRASETPublicationsNo ratings yet
- Microarray Time SeriesDocument19 pagesMicroarray Time SeriesMohamed MahmoudNo ratings yet
- Efficacy of Algorithms in Deep Learning On Brain Tumor Cancer Detection (Topic Area Deep Learning)Document8 pagesEfficacy of Algorithms in Deep Learning On Brain Tumor Cancer Detection (Topic Area Deep Learning)International Journal of Innovative Science and Research TechnologyNo ratings yet
- Hybrid Genetic Algorithm-Neural Network: Feature Extraction For Unpreprocessed Microarray DataDocument10 pagesHybrid Genetic Algorithm-Neural Network: Feature Extraction For Unpreprocessed Microarray DataTajbia HossainNo ratings yet
- Resolving Gene Expression Data Using Multiobjective Optimization ApproachDocument7 pagesResolving Gene Expression Data Using Multiobjective Optimization ApproachIJIRSTNo ratings yet
- Grid Search-Based Hyperparameter Tuning and Classification of Microarray Cancer DataDocument8 pagesGrid Search-Based Hyperparameter Tuning and Classification of Microarray Cancer DataannalystatNo ratings yet
- Malaria Detection Using Deep Convolutional Neural Networks: ISSN-2394-5125Document9 pagesMalaria Detection Using Deep Convolutional Neural Networks: ISSN-2394-5125GEORGENo ratings yet
- Spaper 4Document11 pagesSpaper 4Rozita JackNo ratings yet
- Review Article: A Review of Feature Selection and Feature Extraction Methods Applied On Microarray DataDocument14 pagesReview Article: A Review of Feature Selection and Feature Extraction Methods Applied On Microarray Datafahad darNo ratings yet
- Microarray Data Analysis and Mining Tools: Bioinformation April 2011Document6 pagesMicroarray Data Analysis and Mining Tools: Bioinformation April 2011Dr. Megha PUNo ratings yet
- Applications of Machine Learning Techniques To Predict Diagnostic Breast CancerDocument11 pagesApplications of Machine Learning Techniques To Predict Diagnostic Breast CancerAkashi DogeyNo ratings yet
- Classification of Cancerous Profiles Using Machine LearningDocument38 pagesClassification of Cancerous Profiles Using Machine LearningRuby IniyaNo ratings yet
- A Study On Deep Learning For Bioinformatics: Panchami - VU, Dr. Hariharan.N, Dr. Manish - TIDocument5 pagesA Study On Deep Learning For Bioinformatics: Panchami - VU, Dr. Hariharan.N, Dr. Manish - TIRahul SharmaNo ratings yet
- Pattern Recognition: V. Bolo N-Canedo, N. Sa Nchez-Maron O, A. Alonso-BetanzosDocument9 pagesPattern Recognition: V. Bolo N-Canedo, N. Sa Nchez-Maron O, A. Alonso-BetanzosTajbia HossainNo ratings yet
- Gene Mutation PDFDocument5 pagesGene Mutation PDFChandan NaiduNo ratings yet
- Research Paper 1 PublicationDocument4 pagesResearch Paper 1 PublicationSri Vidya MakkapatiNo ratings yet
- TS PPT (Autosaved)Document16 pagesTS PPT (Autosaved)Vasiha Fathima RNo ratings yet
- Evolutionary Computational Algorithm by Blending of PPCA and EP-Enhanced Supervised Classifier For Microarray Gene Expression DataDocument10 pagesEvolutionary Computational Algorithm by Blending of PPCA and EP-Enhanced Supervised Classifier For Microarray Gene Expression DataIAES IJAINo ratings yet
- Optimizing Classification Efficiency With Machine Learning Techniques For Pattern MatchingDocument18 pagesOptimizing Classification Efficiency With Machine Learning Techniques For Pattern Matchingm.albaiti111No ratings yet
- 22-GWO Papers-27-02-2024Document16 pages22-GWO Papers-27-02-2024gunjanbhartia2003No ratings yet
- 173 701 1 PBDocument12 pages173 701 1 PBArendraNo ratings yet
- Analytical Study of Diagnosis For Angioplasty and Stents Patients Using Improved Classification TechniqueDocument4 pagesAnalytical Study of Diagnosis For Angioplasty and Stents Patients Using Improved Classification TechniqueEditor IJTSRDNo ratings yet
- Deep Learning Based Convolutional Neural Networks (DLCNN) On Classification Algorithm To Detect The Brain Turnor Diseases Using MRI and CT Scan ImagesDocument8 pagesDeep Learning Based Convolutional Neural Networks (DLCNN) On Classification Algorithm To Detect The Brain Turnor Diseases Using MRI and CT Scan ImagesInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Study On Malaria Detection Using Transfer LearningDocument9 pagesStudy On Malaria Detection Using Transfer LearningIJRASETPublicationsNo ratings yet
- Convolutional Neural Networks Based Plant Leaf Diseases Detection SchemeDocument7 pagesConvolutional Neural Networks Based Plant Leaf Diseases Detection SchemeSudhaNo ratings yet
- A Modified Resnet (MRestNet) Based Classification For Brain Tumor DatasetDocument18 pagesA Modified Resnet (MRestNet) Based Classification For Brain Tumor DatasetVinayaga MoorthyNo ratings yet
- Scmra: A Robust Deep Learning Method To Annotate Scrna-Seq Data With Multiple Reference DatasetsDocument15 pagesScmra: A Robust Deep Learning Method To Annotate Scrna-Seq Data With Multiple Reference DatasetsNavid ArefanNo ratings yet
- Analytical Estimation of Quantum Convolutional Neural Network and Convolutional Neural Network For Breast Cancer DetectionDocument11 pagesAnalytical Estimation of Quantum Convolutional Neural Network and Convolutional Neural Network For Breast Cancer DetectionInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- draftDNAPressChapter v8Document77 pagesdraftDNAPressChapter v8animboyNo ratings yet
- Deep Convolutional Neural Network Framework With Multi-Modal Fusion For Alzheimer's DetectionDocument13 pagesDeep Convolutional Neural Network Framework With Multi-Modal Fusion For Alzheimer's DetectionIJRES teamNo ratings yet
- Jurnal 3Document15 pagesJurnal 3fikaadillah3No ratings yet
- A Deep Neural Network Model Using Random Forest ToDocument9 pagesA Deep Neural Network Model Using Random Forest ToGisselyNo ratings yet
- A Survey in DNA Sequence Classification by Soft Computing MethodsDocument6 pagesA Survey in DNA Sequence Classification by Soft Computing MethodsBackiya Lakshmi KumaresanNo ratings yet
- A Comparative Study of Tuberculosis DetectionDocument5 pagesA Comparative Study of Tuberculosis DetectionMehera Binte MizanNo ratings yet
- A Comparative Study For Brain Tumor Detection Analysis Using CNN and VGG-16 and Its ApplicationDocument8 pagesA Comparative Study For Brain Tumor Detection Analysis Using CNN and VGG-16 and Its ApplicationInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Bio Report ElDocument8 pagesBio Report ElJateen RathodNo ratings yet
- Skin Cancer Classification Using Convolutional Neural NetworksDocument8 pagesSkin Cancer Classification Using Convolutional Neural NetworksNirmalendu KumarNo ratings yet
- Predicting Clinical Outcomes From Genomics Deep Learning ModelsDocument11 pagesPredicting Clinical Outcomes From Genomics Deep Learning ModelsVenkat KunaNo ratings yet
- Rapid Dna Origami Nanostructure Detection and Classification Using The Yolov5 Deep Convolutional Neural NetworkDocument13 pagesRapid Dna Origami Nanostructure Detection and Classification Using The Yolov5 Deep Convolutional Neural NetworkEd swertNo ratings yet
- Analysis of Heart Disease Using in Data Mining Tools Orange and WekaDocument7 pagesAnalysis of Heart Disease Using in Data Mining Tools Orange and WekabekNo ratings yet
- Breast Cancer Detection Using SVM Classifier With Grid Search TechniqueDocument6 pagesBreast Cancer Detection Using SVM Classifier With Grid Search TechniqueroniwahyuNo ratings yet
- CNN-Based Image Analysis For Malaria Diagnosis: Abstract - Malaria Is A Major Global Health Threat. TheDocument4 pagesCNN-Based Image Analysis For Malaria Diagnosis: Abstract - Malaria Is A Major Global Health Threat. TheMiftahul RakaNo ratings yet
- Project (Sec: 01)Document10 pagesProject (Sec: 01)Amanat Khan ShishirNo ratings yet
- DATA MINING and MACHINE LEARNING. CLASSIFICATION PREDICTIVE TECHNIQUES: NAIVE BAYES, NEAREST NEIGHBORS and NEURAL NETWORKS: Examples with MATLABFrom EverandDATA MINING and MACHINE LEARNING. CLASSIFICATION PREDICTIVE TECHNIQUES: NAIVE BAYES, NEAREST NEIGHBORS and NEURAL NETWORKS: Examples with MATLABNo ratings yet
- Innovative Mathematical Insights through Artificial Intelligence (AI): Analysing Ramanujan Series and the Relationship between e and π\piDocument8 pagesInnovative Mathematical Insights through Artificial Intelligence (AI): Analysing Ramanujan Series and the Relationship between e and π\piInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Intrusion Detection and Prevention Systems for Ad-Hoc NetworksDocument8 pagesIntrusion Detection and Prevention Systems for Ad-Hoc NetworksInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- AI Robots in Various SectorDocument3 pagesAI Robots in Various SectorInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- An Explanatory Sequential Study of Public Elementary School Teachers on Deped Computerization Program (DCP)Document7 pagesAn Explanatory Sequential Study of Public Elementary School Teachers on Deped Computerization Program (DCP)International Journal of Innovative Science and Research TechnologyNo ratings yet
- Nurturing Corporate Employee Emotional Wellbeing, Time Management and the Influence on FamilyDocument8 pagesNurturing Corporate Employee Emotional Wellbeing, Time Management and the Influence on FamilyInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Fuzzy based Tie-Line and LFC of a Two-Area Interconnected SystemDocument6 pagesFuzzy based Tie-Line and LFC of a Two-Area Interconnected SystemInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Dynamic Analysis of High-Rise Buildings for Various Irregularities with and without Floating ColumnDocument3 pagesDynamic Analysis of High-Rise Buildings for Various Irregularities with and without Floating ColumnInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Importance of Early Intervention of Traumatic Cataract in ChildrenDocument5 pagesImportance of Early Intervention of Traumatic Cataract in ChildrenInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Geotechnical Assessment of Selected Lateritic Soils in Southwest Nigeria for Road Construction and Development of Artificial Neural Network Mathematical Based Model for Prediction of the California Bearing RatioDocument10 pagesGeotechnical Assessment of Selected Lateritic Soils in Southwest Nigeria for Road Construction and Development of Artificial Neural Network Mathematical Based Model for Prediction of the California Bearing RatioInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- The Effects of Liquid Density and Impeller Size with Volute Clearance on the Performance of Radial Blade Centrifugal Pumps: An Experimental ApproachDocument16 pagesThe Effects of Liquid Density and Impeller Size with Volute Clearance on the Performance of Radial Blade Centrifugal Pumps: An Experimental ApproachInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Integration of Information Communication Technology, Strategic Leadership and Academic Performance in Universities in North Kivu, Democratic Republic of CongoDocument7 pagesIntegration of Information Communication Technology, Strategic Leadership and Academic Performance in Universities in North Kivu, Democratic Republic of CongoInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- A Novel Approach to Template Filling with Automatic Speech Recognition for Healthcare ProfessionalsDocument6 pagesA Novel Approach to Template Filling with Automatic Speech Recognition for Healthcare ProfessionalsInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Breaking Down Barriers To Inclusion: Stories of Grade Four Teachers in Maintream ClassroomsDocument14 pagesBreaking Down Barriers To Inclusion: Stories of Grade Four Teachers in Maintream ClassroomsInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- The Influence of Artificial Intelligence On Employment Trends in The United States (US)Document5 pagesThe Influence of Artificial Intelligence On Employment Trends in The United States (US)International Journal of Innovative Science and Research TechnologyNo ratings yet
- Diode Laser Therapy for Drug Induced Gingival Enlaregement: A CaseReportDocument4 pagesDiode Laser Therapy for Drug Induced Gingival Enlaregement: A CaseReportInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Comparison of JavaScript Frontend Frameworks - Angular, React, and VueDocument8 pagesComparison of JavaScript Frontend Frameworks - Angular, React, and VueInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Analysis of Factors Obstacling Construction Work in The Tojo Una-Una Islands RegionDocument9 pagesAnalysis of Factors Obstacling Construction Work in The Tojo Una-Una Islands RegionInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Occupational Safety: PPE Use and Hazard Experiences Among Welders in Valencia City, BukidnonDocument11 pagesOccupational Safety: PPE Use and Hazard Experiences Among Welders in Valencia City, BukidnonInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Organizational Factors That Influence Information Security in Smes: A Case Study of Mogadishu, SomaliaDocument10 pagesOrganizational Factors That Influence Information Security in Smes: A Case Study of Mogadishu, SomaliaInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Disseminating The Real-World Importance of Conjunct Studies of Acculturation, Transculturation, and Deculturation Processes: Why This Can Be A Useful Technique To Analyze Real-World ObservationsDocument15 pagesDisseminating The Real-World Importance of Conjunct Studies of Acculturation, Transculturation, and Deculturation Processes: Why This Can Be A Useful Technique To Analyze Real-World ObservationsInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Mediating Effect of Encouraging Attitude of School Principals On Personal Well-Being and Career Ethics of TeachersDocument10 pagesMediating Effect of Encouraging Attitude of School Principals On Personal Well-Being and Career Ethics of TeachersInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- The Relationship of Total Quality Management Practices and Project Performance With Risk Management As Mediator: A Study of East Coast Rail Link Project in MalaysiaDocument19 pagesThe Relationship of Total Quality Management Practices and Project Performance With Risk Management As Mediator: A Study of East Coast Rail Link Project in MalaysiaInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Classifying Crop Leaf Diseases Using Different Deep Learning Models With Transfer LearningDocument8 pagesClassifying Crop Leaf Diseases Using Different Deep Learning Models With Transfer LearningInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- The Relationship Between Knowledge About Halitosis and Dental and Oral Hygiene in Patients With Fixed Orthodontics at The HK Medical Center MakassarDocument4 pagesThe Relationship Between Knowledge About Halitosis and Dental and Oral Hygiene in Patients With Fixed Orthodontics at The HK Medical Center MakassarInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Pecha Kucha Presentation in The University English Classes: Advantages and DisadvantagesDocument4 pagesPecha Kucha Presentation in The University English Classes: Advantages and DisadvantagesInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Total Cost of Ownership of Electric Car and Internal Combustion Engine Car With Performance NormalizationDocument11 pagesTotal Cost of Ownership of Electric Car and Internal Combustion Engine Car With Performance NormalizationInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Integrating Multimodal Deep Learning For Enhanced News Sentiment Analysis and Market Movement ForecastingDocument8 pagesIntegrating Multimodal Deep Learning For Enhanced News Sentiment Analysis and Market Movement ForecastingInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- An Investigation Model To Combat Computer Gaming Delinquency in South Africa, Server-Based Gaming, and Illegal Online GamingDocument15 pagesAn Investigation Model To Combat Computer Gaming Delinquency in South Africa, Server-Based Gaming, and Illegal Online GamingInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Gender-Based Violence: Engaging Children in The SolutionDocument10 pagesGender-Based Violence: Engaging Children in The SolutionInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Development of Creative Thinking: Case Study of Basic Design StudioDocument9 pagesDevelopment of Creative Thinking: Case Study of Basic Design StudioInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- PDF Deep Reinforcement Learning in Action 1St Edition Alexander Zai Ebook Full ChapterDocument53 pagesPDF Deep Reinforcement Learning in Action 1St Edition Alexander Zai Ebook Full Chapterrick.kelleher522100% (3)
- Multimae: Multi-Modal Multi-Task Masked AutoencodersDocument21 pagesMultimae: Multi-Modal Multi-Task Masked AutoencodersAnya PankovaNo ratings yet
- Artificial Intelligence in Food Industry: Submitted To: Sir Anwar Submitted By: Gul E Kainat 15-ARID-4939Document32 pagesArtificial Intelligence in Food Industry: Submitted To: Sir Anwar Submitted By: Gul E Kainat 15-ARID-4939Lubna KhanNo ratings yet
- How To Use Google Colaboratory For Video Processing - CodeProjectDocument12 pagesHow To Use Google Colaboratory For Video Processing - CodeProjectgfgomesNo ratings yet
- Play With Data ScienceDocument306 pagesPlay With Data SciencedpsharmaNo ratings yet
- ML - Expectation-Maximization AlgorithmDocument3 pagesML - Expectation-Maximization AlgorithmprakashNo ratings yet
- CART v6.0Document434 pagesCART v6.0segorin2No ratings yet
- Artificial Intelligence Machine Learning Program BrochureDocument24 pagesArtificial Intelligence Machine Learning Program BrochureSunnyNo ratings yet
- Deep Learning For Prediction of Depressive Symptoms in A Large Textual DatasetDocument24 pagesDeep Learning For Prediction of Depressive Symptoms in A Large Textual DatasetAmith PrasanthNo ratings yet
- Optimization in Machine LearningDocument26 pagesOptimization in Machine LearningrbernalMxNo ratings yet
- 07 ML Classificaion Advanced KappaDocument18 pages07 ML Classificaion Advanced KappaIn TechNo ratings yet
- B.tech 20-21 InternshipDocument9 pagesB.tech 20-21 Internship20501A05D5 NASIKA PRAVALLIKANo ratings yet
- Keystroke DynamicsDocument6 pagesKeystroke DynamicsrasbiakNo ratings yet
- Dissertation Research Aims and ObjectivesDocument7 pagesDissertation Research Aims and ObjectivesHelpWritingACollegePaperCanada100% (1)
- Book Review:: Pedro Domingos. Basic Books. 2015. ISBN 978-0465065707Document2 pagesBook Review:: Pedro Domingos. Basic Books. 2015. ISBN 978-0465065707Anurag GuptaNo ratings yet
- A Paper On Stylometric Differences Between Authentic and Fabricated AhadithDocument18 pagesA Paper On Stylometric Differences Between Authentic and Fabricated AhadithMuhammad Ammar adilNo ratings yet
- Modeling PROTAC Degradation Activity With Machine LearningDocument12 pagesModeling PROTAC Degradation Activity With Machine Learningpradeep kumarNo ratings yet
- Written Work 1 - Attempt Review3Document6 pagesWritten Work 1 - Attempt Review3RussianOmeleteNo ratings yet
- Machine Learning Made Easy: A Review of Scikit-Learn Package in Python Programming LanguageDocument14 pagesMachine Learning Made Easy: A Review of Scikit-Learn Package in Python Programming Languagejuan carlosNo ratings yet
- Deep Learning - Lesson PlanDocument5 pagesDeep Learning - Lesson PlanAnonymous xMYE0TiNBcNo ratings yet
- Machine Learning Algorithms For Signal and Image Processing Suman Lata Tripathi Full ChapterDocument67 pagesMachine Learning Algorithms For Signal and Image Processing Suman Lata Tripathi Full Chapterdolores.godfrey250100% (7)
- Machine Learning PaperDocument5 pagesMachine Learning PaperAshutoshNo ratings yet
- 2022 - Predicting Tunnel Squeezing Using Support Vector Machine Optimized by Whale Optimization AlgorithmDocument24 pages2022 - Predicting Tunnel Squeezing Using Support Vector Machine Optimized by Whale Optimization Algorithm周牮No ratings yet
- Prediction of Reservoir Brittleness From Geophysical Logs Using Machine Learning AlgorithmsDocument16 pagesPrediction of Reservoir Brittleness From Geophysical Logs Using Machine Learning Algorithmsdobeng147No ratings yet
- Deep Face Recognition: A Survey: Mei Wang, Weihong DengDocument31 pagesDeep Face Recognition: A Survey: Mei Wang, Weihong DengGitaNo ratings yet
- ThoughtSpot SpotIQ AI Driven Analytics White Paper PDFDocument20 pagesThoughtSpot SpotIQ AI Driven Analytics White Paper PDFfoliox100% (1)
- Lecture 1 - Introduction To MLDocument25 pagesLecture 1 - Introduction To MLsba20050No ratings yet
- Machine Learning Using Python - Discover The World of MLDocument108 pagesMachine Learning Using Python - Discover The World of MLbahiniy286No ratings yet