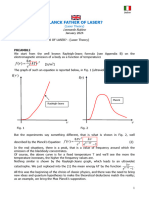
Download as pdf or txt
You might also like
- Fermi-Dirac Statistics 1Document27 pagesFermi-Dirac Statistics 1lucky jNo ratings yet
- Math 139 Fourier Analysis Notes PDFDocument212 pagesMath 139 Fourier Analysis Notes PDFAidan HolwerdaNo ratings yet
- Force Autocorrelation Function in Linear Response Theory and The Origin of FrictionDocument24 pagesForce Autocorrelation Function in Linear Response Theory and The Origin of Frictionadil kaplanserenNo ratings yet
- Valence: Integral, Mixed, Nearly Integral: Fi (?fi ($)Document1 pageValence: Integral, Mixed, Nearly Integral: Fi (?fi ($)SupriyaNo ratings yet
- (George v. Bassis) Kms-ConditionDocument12 pages(George v. Bassis) Kms-ConditionLUIS QUEZADANo ratings yet
- Encounters With The Golden Ratio in Fluid Dynamics: M. MokryDocument10 pagesEncounters With The Golden Ratio in Fluid Dynamics: M. MokryLeon BlažinovićNo ratings yet
- 1949 - Rate of Nucleation of Condense Systems (Fischer Turnbull)Document4 pages1949 - Rate of Nucleation of Condense Systems (Fischer Turnbull)SoodooNavindraNo ratings yet
- LevyDocument4 pagesLevyRamon FerreiraNo ratings yet
- 8 - Random Walk - 1974 - A Course in Probability TheoryDocument45 pages8 - Random Walk - 1974 - A Course in Probability TheoryGramm ChaoNo ratings yet
- Fermi-Dirac Distribution and The Fermi-LevelDocument27 pagesFermi-Dirac Distribution and The Fermi-Levelsudhanshu kumar50% (2)
- 03 Equilibria (I)Document11 pages03 Equilibria (I)David LevisteNo ratings yet
- History of IntegralDocument9 pagesHistory of IntegralYusrina Nur AmaliaNo ratings yet
- The Fresnel Equations and Brewster's Law: EquipmentDocument6 pagesThe Fresnel Equations and Brewster's Law: EquipmentDilip Sahu100% (1)
- Set of Questions 10Document1 pageSet of Questions 10ApuNo ratings yet
- Sheet 09 SolutionDocument7 pagesSheet 09 SolutionDaniel PradoNo ratings yet
- Streda+Integers v2Document5 pagesStreda+Integers v2Kariem Mohamed Ragab HamedNo ratings yet
- Electromagnetic Waves - 2Document15 pagesElectromagnetic Waves - 2verdy rahadianNo ratings yet
- Planck Father of Laser - Laser Theory - Eng+ItaDocument26 pagesPlanck Father of Laser - Laser Theory - Eng+ItaLeonardo RubinoNo ratings yet
- VB Bhatia 1Document25 pagesVB Bhatia 1Ammu k67% (3)
- B 2 Ps 3Document4 pagesB 2 Ps 3Sifei ZhangNo ratings yet
- Potential Vorticity: DerivationDocument5 pagesPotential Vorticity: DerivationSteven ScottNo ratings yet
- Kirchberg's TheoremDocument8 pagesKirchberg's TheoremDragan RukavinaNo ratings yet
- PresentationDocument19 pagesPresentationBilal HameedNo ratings yet
- Lecture 9 - Anharmonic Effects in CrystalsDocument19 pagesLecture 9 - Anharmonic Effects in Crystalsanbarasanr29102001No ratings yet
- Chap 17Document7 pagesChap 17apratim.chatterjiNo ratings yet
- Density Functionals For Couiomb Systems: Departments New Jersey 08544, U.S.ADocument35 pagesDensity Functionals For Couiomb Systems: Departments New Jersey 08544, U.S.ARamon FerreiraNo ratings yet
- Finite de Finetti Style THRMDocument15 pagesFinite de Finetti Style THRMtrump bartholomewNo ratings yet
- As A A At: 11.3 Kondo Eo (J) /LCDocument1 pageAs A A At: 11.3 Kondo Eo (J) /LCSupriyaNo ratings yet
- Susceptibility: General Approach Perturbation Theoretic Calculations of Nonlinear Coefficient For Ferroelectric MaterialsDocument3 pagesSusceptibility: General Approach Perturbation Theoretic Calculations of Nonlinear Coefficient For Ferroelectric Materialsrongo024No ratings yet
- All-Or-None (Or Something in Between) Transition of DNA: Europhys. Lett.Document7 pagesAll-Or-None (Or Something in Between) Transition of DNA: Europhys. Lett.JasonNo ratings yet
- Encounters With The Golden Ratio in Fluid DynamicsDocument11 pagesEncounters With The Golden Ratio in Fluid DynamicsDeepak Kumar Singh Res. Scholar., Dept. of Mechanical Engg., IIT (BHU)No ratings yet
- Paper2 Influence Variable Surface Tension Capillary GravityDocument14 pagesPaper2 Influence Variable Surface Tension Capillary Gravityersin_ozugurluNo ratings yet
- A F Nan ScatteringDocument32 pagesA F Nan ScatteringkrishnaNo ratings yet
- Lattice Gas Cellular Automata and Lattice Boltzmann Models Chapter6Document88 pagesLattice Gas Cellular Automata and Lattice Boltzmann Models Chapter6api-3702465No ratings yet
- A Study of Magnetic ViscosityDocument12 pagesA Study of Magnetic ViscositySantiago BNo ratings yet
- Degree of Local Zeta Functions and MonodromyDocument11 pagesDegree of Local Zeta Functions and MonodromyJan DenefNo ratings yet
- HW9Document3 pagesHW9Panneer SelvamNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L3Document5 pagesStatistical Mechanics Lecture Notes (2006), L3OmegaUserNo ratings yet
- 6CCP3212 Statistical Mechanics Homework 2Document4 pages6CCP3212 Statistical Mechanics Homework 2Hameed ul haqNo ratings yet
- Connections Chapter4Document20 pagesConnections Chapter4Keeley HoekNo ratings yet
- Unit 10Document16 pagesUnit 10airtelbanikoct2023No ratings yet
- Chap 11Document16 pagesChap 11selinnertas1No ratings yet
- Tut 3Document2 pagesTut 3Akriti SinghNo ratings yet
- Kraus OpsDocument13 pagesKraus OpsHarmanjot SinghNo ratings yet
- APshock MethodsDocument22 pagesAPshock MethodsRoy VeseyNo ratings yet
- A Tauberian Theorem For A Class of Function Spaces: Miguel A. Jim EnezDocument9 pagesA Tauberian Theorem For A Class of Function Spaces: Miguel A. Jim EnezBrandon WarrenNo ratings yet
- Complex WavesDocument4 pagesComplex WavesGodwin LarryNo ratings yet
- Solutions To Problem Set 4: ConnectednessDocument5 pagesSolutions To Problem Set 4: ConnectednessDaniel E. Quintero PamplonaNo ratings yet
- Assignment 2Document6 pagesAssignment 2Keshav SinglaNo ratings yet
- Parameters of Bouc-Wen Hysteretic Model RevisitedDocument8 pagesParameters of Bouc-Wen Hysteretic Model RevisitedFoudilYouyouNo ratings yet
- Baire Classification of I-Density and I-Approximately Continuous FunctionsDocument17 pagesBaire Classification of I-Density and I-Approximately Continuous FunctionsandreNo ratings yet
- Carlde Boor': Journal of Approximation Theory 1, 219-235Document17 pagesCarlde Boor': Journal of Approximation Theory 1, 219-235lukkioNo ratings yet
- Ps 9Document2 pagesPs 9CLERK SULCA QUISPENo ratings yet
- Spectrum of /-Fluids: A Statistical Derivation: J. A. E. Carrulo, T J. A. S. Lima, 2A and A. Maia, Jr. 4Document6 pagesSpectrum of /-Fluids: A Statistical Derivation: J. A. E. Carrulo, T J. A. S. Lima, 2A and A. Maia, Jr. 4wintersoulsNo ratings yet
- 10 Electric CurrentDocument12 pages10 Electric Currentömer aydınNo ratings yet
- The Voltage of The PhotonDocument15 pagesThe Voltage of The PhotonFB100% (1)
- 9205 Chap01Document18 pages9205 Chap01Shridhar MathadNo ratings yet
- The Equidistribution Theory of Holomorphic Curves. (AM-64), Volume 64From EverandThe Equidistribution Theory of Holomorphic Curves. (AM-64), Volume 64No ratings yet
- MovingCharges&Magnetism 03-1-15PMDocument15 pagesMovingCharges&Magnetism 03-1-15PMNeeraj MeenaNo ratings yet
- Doubt Clearing Session With Anno 1697607249646Document174 pagesDoubt Clearing Session With Anno 1697607249646Neeraj MeenaNo ratings yet
- Tricks For Parity and Expectations With Anno 1697607254597Document65 pagesTricks For Parity and Expectations With Anno 1697607254597Neeraj MeenaNo ratings yet
- UOK-Ph.D. Fee StructureDocument2 pagesUOK-Ph.D. Fee StructureNeeraj MeenaNo ratings yet
- 1617 - UOK-Registered Research Supervisors-Faculty of ScienceDocument3 pages1617 - UOK-Registered Research Supervisors-Faculty of ScienceNeeraj MeenaNo ratings yet
- Lecture Notes On Electric FieldDocument4 pagesLecture Notes On Electric FieldRenjith Raveendran Pillai100% (1)
- Electrostatics: Electric Field & Potential: Example 1: Two Intersecting and Oppositely Charged SpheresDocument11 pagesElectrostatics: Electric Field & Potential: Example 1: Two Intersecting and Oppositely Charged SpheresgopalNo ratings yet
- Systematic Errors in The ACME Electron EDM Experiment: e e e eDocument9 pagesSystematic Errors in The ACME Electron EDM Experiment: e e e eDaniel Gordon AngNo ratings yet
- Electromagnetism Top 500 Question Bank For JEE Main by MathonGoDocument59 pagesElectromagnetism Top 500 Question Bank For JEE Main by MathonGopratikpotadar124No ratings yet
- 古典電動力學題庫Document14 pages古典電動力學題庫joshua7312No ratings yet
- CH 4 PhyDocument38 pagesCH 4 PhyMalikNo ratings yet
- Elec P CDocument31 pagesElec P CKria Vora100% (1)
- 1797 PhysicsxiiDocument115 pages1797 Physicsxiiabhijit gogoiNo ratings yet
- 1.electric Charge & Fields - TestDocument18 pages1.electric Charge & Fields - TestArshdeep singhNo ratings yet
- Xii Physics Chapter1-BookDocument25 pagesXii Physics Chapter1-BookChitra KaruppiahNo ratings yet
- ElectromagneticsDocument170 pagesElectromagneticsMina Riad AboudNo ratings yet
- (Physics) R Gupta's Popular Master Guide 2022Document150 pages(Physics) R Gupta's Popular Master Guide 2022beniiiNo ratings yet
- CBSE Class 12 Physics Current ElectricityDocument21 pagesCBSE Class 12 Physics Current ElectricityK_S_Krishna0001No ratings yet
- Objective Questions: Rubbed With MadeDocument12 pagesObjective Questions: Rubbed With MadeANIKET ROYNo ratings yet
- Emft NotesDocument17 pagesEmft Notesvinodkumarsingh1985No ratings yet
- CH 21Document37 pagesCH 21al hNo ratings yet
- 3 - BK Modified Electrical PropertiesDocument120 pages3 - BK Modified Electrical PropertiesRam Kiran DevireddyNo ratings yet
- Radiofrequency Radiation Dosimetry HandbookDocument330 pagesRadiofrequency Radiation Dosimetry Handbookle84No ratings yet
- Key Points: Unit I - IIDocument10 pagesKey Points: Unit I - IINarendra SNo ratings yet
- Physics Holidays HWDocument16 pagesPhysics Holidays HWvirender singhNo ratings yet
- UNIT 1 CHEMISTRyDocument101 pagesUNIT 1 CHEMISTRyRishabh KothariNo ratings yet
- Phy M.C.QDocument66 pagesPhy M.C.QmanasNo ratings yet
- Xii ScienceDocument23 pagesXii Scienceom balaNo ratings yet
- Resonance in Physics, Chemistry and Biology.: January 2001Document62 pagesResonance in Physics, Chemistry and Biology.: January 2001binodeNo ratings yet
- Namma Kalvi 12th Physics Model Question Papers With Answers em 220129Document158 pagesNamma Kalvi 12th Physics Model Question Papers With Answers em 220129karthi1234No ratings yet
- EM LectureNoteDocument90 pagesEM LectureNoteQuỳnh GiaoNo ratings yet
- Bor Kavcic: Electrodynamics of Human HeartDocument14 pagesBor Kavcic: Electrodynamics of Human HearttohunabohunNo ratings yet
- Assignment 1 PDFDocument46 pagesAssignment 1 PDFTechnical Raj100% (1)
- Ilovepdf MergedDocument21 pagesIlovepdf MergedraghavchawdhryNo ratings yet
- Principles of Electrodynamics BY MILES MATHISDocument10 pagesPrinciples of Electrodynamics BY MILES MATHISlennysanchezNo ratings yet