
Gnipst 4 30 5694 2.4
Gnipst 4 30 5694 2.4
You might also like
- Annex 7: WHO Guidelines On Transfer of Technology in Pharmaceutical ManufacturingDocument25 pagesAnnex 7: WHO Guidelines On Transfer of Technology in Pharmaceutical ManufacturingEkin Duran100% (1)
- Kit Kat Case StudyDocument6 pagesKit Kat Case Studyms.Ahmed0% (1)
- Lab 2Document30 pagesLab 2jianneNo ratings yet
- SOP For Continued Process VerificationDocument9 pagesSOP For Continued Process VerificationMubarak Patel100% (1)
- Technology Transfer in Pharmaceutical IndustryDocument24 pagesTechnology Transfer in Pharmaceutical IndustryPranjal100% (3)
- Parametric ReleaseDocument7 pagesParametric ReleaseshdphNo ratings yet
- Risk Identification For Aseptic ProcessingDocument42 pagesRisk Identification For Aseptic Processingmmmmm100% (2)
- Validation and Validation ProtocolDocument18 pagesValidation and Validation ProtocolLalit Lata JhaNo ratings yet
- Unit - Granularity Technology Transfer - SVKDocument167 pagesUnit - Granularity Technology Transfer - SVKHarshada AgarkarNo ratings yet
- Peer-Reviewed: Medical Devices: Warning LettersDocument1 pagePeer-Reviewed: Medical Devices: Warning LettersSandra RodriguezNo ratings yet
- Guidance For Industry: Process ValidationDocument18 pagesGuidance For Industry: Process ValidationBruno DebonnetNo ratings yet
- Technology Transfer V.1.0Document39 pagesTechnology Transfer V.1.0Syahid AchyarNo ratings yet
- DocumentationDocument46 pagesDocumentationajak16406No ratings yet
- TRS961 - Annex7 WHO Tech TransferDocument25 pagesTRS961 - Annex7 WHO Tech TransferkrasataNo ratings yet
- ISO 11607 PackagingDocument8 pagesISO 11607 PackagingAngel CvetanovNo ratings yet
- Gnipst 4 30 5694 2.1Document5 pagesGnipst 4 30 5694 2.1Antar GayenNo ratings yet
- Comments On Ongoing Process VerifDocument2 pagesComments On Ongoing Process VerifsofianesedkaouiNo ratings yet
- Gnipst 4 30 5694 2.2Document2 pagesGnipst 4 30 5694 2.2Antar GayenNo ratings yet
- ValidationDocument49 pagesValidationAshokPokiriNo ratings yet
- USP-Lifecycle Management of Analytical ProceduresDocument15 pagesUSP-Lifecycle Management of Analytical ProceduresNur AcarNo ratings yet
- Articulo Tecnologia de TransferenciaDocument10 pagesArticulo Tecnologia de Transferencia20172504No ratings yet
- TRS961 Annex7Document25 pagesTRS961 Annex7Tahir KhanNo ratings yet
- ValidationDocument5 pagesValidationjyothisahadevanNo ratings yet
- VAL MANUAL 018 Potential Critical Packaging Process Parameters and Validation Practices SampleDocument3 pagesVAL MANUAL 018 Potential Critical Packaging Process Parameters and Validation Practices SampleRahul VermaNo ratings yet
- 2016Document40 pages2016schumonNo ratings yet
- Chinna Nagamalleswara Reddy Alla: SummaryDocument5 pagesChinna Nagamalleswara Reddy Alla: SummaryVijay LS SolutionsNo ratings yet
- RequestDocument3 pagesRequestiloveit52252No ratings yet
- Pharmaceutical Development Q8 (R2) : International Conference On Harmonisation (ICH)Document11 pagesPharmaceutical Development Q8 (R2) : International Conference On Harmonisation (ICH)Md. Akil Mahmud100% (1)
- Gnipst 4 30 5694 2.5Document4 pagesGnipst 4 30 5694 2.5Antar GayenNo ratings yet
- Types of Process ValidationDocument5 pagesTypes of Process ValidationLuanne Dela CruzNo ratings yet
- Technology Transfer - 20230918 - 201200 - 0000Document9 pagesTechnology Transfer - 20230918 - 201200 - 0000KALP PATELNo ratings yet
- Process Analytical TechnologyDocument57 pagesProcess Analytical Technologycarleen_almiraNo ratings yet
- Process-Validation - General-Principles-and-Practices-10Document1 pageProcess-Validation - General-Principles-and-Practices-10Kendry JoseNo ratings yet
- CLAUSE 8.5 Production and Service ProvisionDocument10 pagesCLAUSE 8.5 Production and Service ProvisionNavnath TamhaneNo ratings yet
- 4.1.5 The Quality Assurance ManagerDocument1 page4.1.5 The Quality Assurance ManagerthisiscookingNo ratings yet
- Documentation: Good Manufacturing Practices Modul-3Document29 pagesDocumentation: Good Manufacturing Practices Modul-3ChristinaNo ratings yet
- Taticek-Product Monitoring & Post-Approval Lifecycle Management of Biotech ProductsDocument36 pagesTaticek-Product Monitoring & Post-Approval Lifecycle Management of Biotech Products刘朝阳No ratings yet
- M4 - Lesson 4 - Continued Process VerificationDocument1 pageM4 - Lesson 4 - Continued Process VerificationWilliam DC RiveraNo ratings yet
- Prospective, Concurrent and Retrospective Validation-GOOD ARTICLEDocument8 pagesProspective, Concurrent and Retrospective Validation-GOOD ARTICLEraju1559405No ratings yet
- Adopting The Product Lifecycle ApproachDocument4 pagesAdopting The Product Lifecycle Approach刘朝阳No ratings yet
- DR KyQUALITY by DESIGN:From Theory To PracticeDocument25 pagesDR KyQUALITY by DESIGN:From Theory To PracticemanipallavanNo ratings yet
- Technology Transfer - 20230927 - 114610 - 0000Document10 pagesTechnology Transfer - 20230927 - 114610 - 0000KALP PATELNo ratings yet
- Qualification Existing Equipment FinalDocument16 pagesQualification Existing Equipment FinalAjay Kumar100% (1)
- 3-2. Assessing Production Documents:: Executed and Master RecordsDocument48 pages3-2. Assessing Production Documents:: Executed and Master RecordsAmer RahmahNo ratings yet
- ValidationDocument49 pagesValidationSwathi Battula100% (1)
- Validation of Pharmaceutical PackagingDocument28 pagesValidation of Pharmaceutical PackagingNaheed Malik100% (3)
- Role and Responsibility of Pharmaceutical Industry Plant PersonnelFrom EverandRole and Responsibility of Pharmaceutical Industry Plant PersonnelNo ratings yet
- Informe 40Document104 pagesInforme 40HOSMANECHEVERRIANo ratings yet
- Annex 4 Supplementary Guidelines On GoodDocument72 pagesAnnex 4 Supplementary Guidelines On GoodHiếu Ngô QuangNo ratings yet
- (920 923) Jul Aug14Document4 pages(920 923) Jul Aug14surafelNo ratings yet
- ASEAN PV (Version 3 1) Includes All AnnexesDocument39 pagesASEAN PV (Version 3 1) Includes All AnnexesAndy RojasNo ratings yet
- Basic Principles of GMP Basic Principles of GMPDocument33 pagesBasic Principles of GMP Basic Principles of GMPTueNo ratings yet
- CGMP GuidelinesDocument23 pagesCGMP GuidelinesRyan 1112No ratings yet
- M4 - Lesson 1 - Introduction To Process ValidationDocument4 pagesM4 - Lesson 1 - Introduction To Process ValidationWilliam DC RiveraNo ratings yet
- Drugs and Health Products: Validation Guidelines For Pharmaceutical Dosage Forms (GUIDE-0029)Document12 pagesDrugs and Health Products: Validation Guidelines For Pharmaceutical Dosage Forms (GUIDE-0029)upadhyayparag01No ratings yet
- 3-2. Assessing Production DocumentsDocument48 pages3-2. Assessing Production DocumentsSandeep sharmaNo ratings yet
- Basic Principles of GMP Basic Principles of GMPDocument33 pagesBasic Principles of GMP Basic Principles of GMPTahir KhanNo ratings yet
- Validation: Presented To: Prof. H.S. Keerthy Department of Pharmaceutics Mallige College of PharmacyDocument26 pagesValidation: Presented To: Prof. H.S. Keerthy Department of Pharmaceutics Mallige College of PharmacyAfdal Naim100% (1)
- Contribute To The Development of A Strategic Plan ILM Assessment Guidance (ML45)Document4 pagesContribute To The Development of A Strategic Plan ILM Assessment Guidance (ML45)Mk Seifu EthioNo ratings yet
- Nursery Car Seat Supplement 2023Document40 pagesNursery Car Seat Supplement 2023doniNo ratings yet
- KP53V85 Tech ManualDocument106 pagesKP53V85 Tech ManualrichiegranNo ratings yet
- UK Wooden Pallet 1200 X 1000mmDocument1 pageUK Wooden Pallet 1200 X 1000mmRizwan rizviNo ratings yet
- A. B. C. D. E.: IIA Report Logistics IndustryDocument14 pagesA. B. C. D. E.: IIA Report Logistics IndustryABHISHEK BHARDWAJNo ratings yet
- Business Plan Hindi Pa FinalDocument10 pagesBusiness Plan Hindi Pa FinalMaria Theresa Cortez MendozaNo ratings yet
- Financial Status and Academic AchievementDocument20 pagesFinancial Status and Academic AchievementkalbokalapatoNo ratings yet
- Logic Controller: Multitechnology Know-HowDocument12 pagesLogic Controller: Multitechnology Know-HowPrIsmatIIcONo ratings yet
- Pipe Flange FormulaDocument57 pagesPipe Flange FormulaRiandi HartartoNo ratings yet
- ABLE Contract Approval.Document5 pagesABLE Contract Approval.Ferris FerrisNo ratings yet
- Tobacco IndustryDocument9 pagesTobacco IndustryjoséNo ratings yet
- TERPS PANS-OPS Approach Brief With Circling Considerations - NBAADocument39 pagesTERPS PANS-OPS Approach Brief With Circling Considerations - NBAAMos DetNo ratings yet
- Accounting Information SystemsDocument11 pagesAccounting Information SystemsMohamed ZakyNo ratings yet
- Social Responsibility Theory FixDocument8 pagesSocial Responsibility Theory Fixsyifa fauziahNo ratings yet
- Absolute Containers Brochure 2019 2 27 PDFDocument19 pagesAbsolute Containers Brochure 2019 2 27 PDFEduardo SolanoNo ratings yet
- Struktur Kelas 10 Ipa 3Document17 pagesStruktur Kelas 10 Ipa 3Okvie AjaNo ratings yet
- Communication Design Insights From The Creative Industries - 2Document33 pagesCommunication Design Insights From The Creative Industries - 2Grace PsycheNo ratings yet
- Hannah Gonzales ResumeDocument2 pagesHannah Gonzales Resumeapi-500481504No ratings yet
- Pig FarmingDocument25 pagesPig Farmingসপ্তক মন্ডল100% (1)
- MZ Instruction and Maintenance ManualDocument25 pagesMZ Instruction and Maintenance ManualLakiLakicNo ratings yet
- Revit-Structural Bim SpecialistDocument5 pagesRevit-Structural Bim SpecialistYoukhanna ZayiaNo ratings yet
- Matrix Korelasi ISO IntergrasiDocument3 pagesMatrix Korelasi ISO IntergrasiAgungMahendraNo ratings yet
- CV Structural EngineerDocument5 pagesCV Structural EngineerMuhammad Ridha100% (1)
- How To Identify Maketing Research ProblemDocument2 pagesHow To Identify Maketing Research ProblempavanNo ratings yet
- Business Schools of AsiaDocument12 pagesBusiness Schools of AsiaRahul JainNo ratings yet
- SGX-Listed Hatten Land Diversifies Beyond Melaka Through Proposed Acquisition of Unicity in Seremban, MalaysiaDocument3 pagesSGX-Listed Hatten Land Diversifies Beyond Melaka Through Proposed Acquisition of Unicity in Seremban, MalaysiaWeR1 Consultants Pte LtdNo ratings yet
- Transmission Hydraulic SystemDocument4 pagesTransmission Hydraulic SystemA Ramos Gaby100% (5)
- AMM - 01-Aug-2019 - 79-21-10-000-004-A - Removal of The Lubrication UnitDocument10 pagesAMM - 01-Aug-2019 - 79-21-10-000-004-A - Removal of The Lubrication UnitIrfan05100% (1)































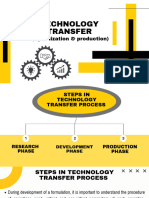










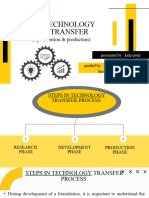















































Download as pdf or txt
You might also like
- Annex 7: WHO Guidelines On Transfer of Technology in Pharmaceutical ManufacturingDocument25 pagesAnnex 7: WHO Guidelines On Transfer of Technology in Pharmaceutical ManufacturingEkin Duran100% (1)
- Kit Kat Case StudyDocument6 pagesKit Kat Case Studyms.Ahmed0% (1)
- Lab 2Document30 pagesLab 2jianneNo ratings yet
- SOP For Continued Process VerificationDocument9 pagesSOP For Continued Process VerificationMubarak Patel100% (1)
- Technology Transfer in Pharmaceutical IndustryDocument24 pagesTechnology Transfer in Pharmaceutical IndustryPranjal100% (3)
- Parametric ReleaseDocument7 pagesParametric ReleaseshdphNo ratings yet
- Risk Identification For Aseptic ProcessingDocument42 pagesRisk Identification For Aseptic Processingmmmmm100% (2)
- Validation and Validation ProtocolDocument18 pagesValidation and Validation ProtocolLalit Lata JhaNo ratings yet
- Unit - Granularity Technology Transfer - SVKDocument167 pagesUnit - Granularity Technology Transfer - SVKHarshada AgarkarNo ratings yet
- Peer-Reviewed: Medical Devices: Warning LettersDocument1 pagePeer-Reviewed: Medical Devices: Warning LettersSandra RodriguezNo ratings yet
- Guidance For Industry: Process ValidationDocument18 pagesGuidance For Industry: Process ValidationBruno DebonnetNo ratings yet
- Technology Transfer V.1.0Document39 pagesTechnology Transfer V.1.0Syahid AchyarNo ratings yet
- DocumentationDocument46 pagesDocumentationajak16406No ratings yet
- TRS961 - Annex7 WHO Tech TransferDocument25 pagesTRS961 - Annex7 WHO Tech TransferkrasataNo ratings yet
- ISO 11607 PackagingDocument8 pagesISO 11607 PackagingAngel CvetanovNo ratings yet
- Gnipst 4 30 5694 2.1Document5 pagesGnipst 4 30 5694 2.1Antar GayenNo ratings yet
- Comments On Ongoing Process VerifDocument2 pagesComments On Ongoing Process VerifsofianesedkaouiNo ratings yet
- Gnipst 4 30 5694 2.2Document2 pagesGnipst 4 30 5694 2.2Antar GayenNo ratings yet
- ValidationDocument49 pagesValidationAshokPokiriNo ratings yet
- USP-Lifecycle Management of Analytical ProceduresDocument15 pagesUSP-Lifecycle Management of Analytical ProceduresNur AcarNo ratings yet
- Articulo Tecnologia de TransferenciaDocument10 pagesArticulo Tecnologia de Transferencia20172504No ratings yet
- TRS961 Annex7Document25 pagesTRS961 Annex7Tahir KhanNo ratings yet
- ValidationDocument5 pagesValidationjyothisahadevanNo ratings yet
- VAL MANUAL 018 Potential Critical Packaging Process Parameters and Validation Practices SampleDocument3 pagesVAL MANUAL 018 Potential Critical Packaging Process Parameters and Validation Practices SampleRahul VermaNo ratings yet
- 2016Document40 pages2016schumonNo ratings yet
- Chinna Nagamalleswara Reddy Alla: SummaryDocument5 pagesChinna Nagamalleswara Reddy Alla: SummaryVijay LS SolutionsNo ratings yet
- RequestDocument3 pagesRequestiloveit52252No ratings yet
- Pharmaceutical Development Q8 (R2) : International Conference On Harmonisation (ICH)Document11 pagesPharmaceutical Development Q8 (R2) : International Conference On Harmonisation (ICH)Md. Akil Mahmud100% (1)
- Gnipst 4 30 5694 2.5Document4 pagesGnipst 4 30 5694 2.5Antar GayenNo ratings yet
- Types of Process ValidationDocument5 pagesTypes of Process ValidationLuanne Dela CruzNo ratings yet
- Technology Transfer - 20230918 - 201200 - 0000Document9 pagesTechnology Transfer - 20230918 - 201200 - 0000KALP PATELNo ratings yet
- Process Analytical TechnologyDocument57 pagesProcess Analytical Technologycarleen_almiraNo ratings yet
- Process-Validation - General-Principles-and-Practices-10Document1 pageProcess-Validation - General-Principles-and-Practices-10Kendry JoseNo ratings yet
- CLAUSE 8.5 Production and Service ProvisionDocument10 pagesCLAUSE 8.5 Production and Service ProvisionNavnath TamhaneNo ratings yet
- 4.1.5 The Quality Assurance ManagerDocument1 page4.1.5 The Quality Assurance ManagerthisiscookingNo ratings yet
- Documentation: Good Manufacturing Practices Modul-3Document29 pagesDocumentation: Good Manufacturing Practices Modul-3ChristinaNo ratings yet
- Taticek-Product Monitoring & Post-Approval Lifecycle Management of Biotech ProductsDocument36 pagesTaticek-Product Monitoring & Post-Approval Lifecycle Management of Biotech Products刘朝阳No ratings yet
- M4 - Lesson 4 - Continued Process VerificationDocument1 pageM4 - Lesson 4 - Continued Process VerificationWilliam DC RiveraNo ratings yet
- Prospective, Concurrent and Retrospective Validation-GOOD ARTICLEDocument8 pagesProspective, Concurrent and Retrospective Validation-GOOD ARTICLEraju1559405No ratings yet
- Adopting The Product Lifecycle ApproachDocument4 pagesAdopting The Product Lifecycle Approach刘朝阳No ratings yet
- DR KyQUALITY by DESIGN:From Theory To PracticeDocument25 pagesDR KyQUALITY by DESIGN:From Theory To PracticemanipallavanNo ratings yet
- Technology Transfer - 20230927 - 114610 - 0000Document10 pagesTechnology Transfer - 20230927 - 114610 - 0000KALP PATELNo ratings yet
- Qualification Existing Equipment FinalDocument16 pagesQualification Existing Equipment FinalAjay Kumar100% (1)
- 3-2. Assessing Production Documents:: Executed and Master RecordsDocument48 pages3-2. Assessing Production Documents:: Executed and Master RecordsAmer RahmahNo ratings yet
- ValidationDocument49 pagesValidationSwathi Battula100% (1)
- Validation of Pharmaceutical PackagingDocument28 pagesValidation of Pharmaceutical PackagingNaheed Malik100% (3)
- Role and Responsibility of Pharmaceutical Industry Plant PersonnelFrom EverandRole and Responsibility of Pharmaceutical Industry Plant PersonnelNo ratings yet
- Informe 40Document104 pagesInforme 40HOSMANECHEVERRIANo ratings yet
- Annex 4 Supplementary Guidelines On GoodDocument72 pagesAnnex 4 Supplementary Guidelines On GoodHiếu Ngô QuangNo ratings yet
- (920 923) Jul Aug14Document4 pages(920 923) Jul Aug14surafelNo ratings yet
- ASEAN PV (Version 3 1) Includes All AnnexesDocument39 pagesASEAN PV (Version 3 1) Includes All AnnexesAndy RojasNo ratings yet
- Basic Principles of GMP Basic Principles of GMPDocument33 pagesBasic Principles of GMP Basic Principles of GMPTueNo ratings yet
- CGMP GuidelinesDocument23 pagesCGMP GuidelinesRyan 1112No ratings yet
- M4 - Lesson 1 - Introduction To Process ValidationDocument4 pagesM4 - Lesson 1 - Introduction To Process ValidationWilliam DC RiveraNo ratings yet
- Drugs and Health Products: Validation Guidelines For Pharmaceutical Dosage Forms (GUIDE-0029)Document12 pagesDrugs and Health Products: Validation Guidelines For Pharmaceutical Dosage Forms (GUIDE-0029)upadhyayparag01No ratings yet
- 3-2. Assessing Production DocumentsDocument48 pages3-2. Assessing Production DocumentsSandeep sharmaNo ratings yet
- Basic Principles of GMP Basic Principles of GMPDocument33 pagesBasic Principles of GMP Basic Principles of GMPTahir KhanNo ratings yet
- Validation: Presented To: Prof. H.S. Keerthy Department of Pharmaceutics Mallige College of PharmacyDocument26 pagesValidation: Presented To: Prof. H.S. Keerthy Department of Pharmaceutics Mallige College of PharmacyAfdal Naim100% (1)
- Contribute To The Development of A Strategic Plan ILM Assessment Guidance (ML45)Document4 pagesContribute To The Development of A Strategic Plan ILM Assessment Guidance (ML45)Mk Seifu EthioNo ratings yet
- Nursery Car Seat Supplement 2023Document40 pagesNursery Car Seat Supplement 2023doniNo ratings yet
- KP53V85 Tech ManualDocument106 pagesKP53V85 Tech ManualrichiegranNo ratings yet
- UK Wooden Pallet 1200 X 1000mmDocument1 pageUK Wooden Pallet 1200 X 1000mmRizwan rizviNo ratings yet
- A. B. C. D. E.: IIA Report Logistics IndustryDocument14 pagesA. B. C. D. E.: IIA Report Logistics IndustryABHISHEK BHARDWAJNo ratings yet
- Business Plan Hindi Pa FinalDocument10 pagesBusiness Plan Hindi Pa FinalMaria Theresa Cortez MendozaNo ratings yet
- Financial Status and Academic AchievementDocument20 pagesFinancial Status and Academic AchievementkalbokalapatoNo ratings yet
- Logic Controller: Multitechnology Know-HowDocument12 pagesLogic Controller: Multitechnology Know-HowPrIsmatIIcONo ratings yet
- Pipe Flange FormulaDocument57 pagesPipe Flange FormulaRiandi HartartoNo ratings yet
- ABLE Contract Approval.Document5 pagesABLE Contract Approval.Ferris FerrisNo ratings yet
- Tobacco IndustryDocument9 pagesTobacco IndustryjoséNo ratings yet
- TERPS PANS-OPS Approach Brief With Circling Considerations - NBAADocument39 pagesTERPS PANS-OPS Approach Brief With Circling Considerations - NBAAMos DetNo ratings yet
- Accounting Information SystemsDocument11 pagesAccounting Information SystemsMohamed ZakyNo ratings yet
- Social Responsibility Theory FixDocument8 pagesSocial Responsibility Theory Fixsyifa fauziahNo ratings yet
- Absolute Containers Brochure 2019 2 27 PDFDocument19 pagesAbsolute Containers Brochure 2019 2 27 PDFEduardo SolanoNo ratings yet
- Struktur Kelas 10 Ipa 3Document17 pagesStruktur Kelas 10 Ipa 3Okvie AjaNo ratings yet
- Communication Design Insights From The Creative Industries - 2Document33 pagesCommunication Design Insights From The Creative Industries - 2Grace PsycheNo ratings yet
- Hannah Gonzales ResumeDocument2 pagesHannah Gonzales Resumeapi-500481504No ratings yet
- Pig FarmingDocument25 pagesPig Farmingসপ্তক মন্ডল100% (1)
- MZ Instruction and Maintenance ManualDocument25 pagesMZ Instruction and Maintenance ManualLakiLakicNo ratings yet
- Revit-Structural Bim SpecialistDocument5 pagesRevit-Structural Bim SpecialistYoukhanna ZayiaNo ratings yet
- Matrix Korelasi ISO IntergrasiDocument3 pagesMatrix Korelasi ISO IntergrasiAgungMahendraNo ratings yet
- CV Structural EngineerDocument5 pagesCV Structural EngineerMuhammad Ridha100% (1)
- How To Identify Maketing Research ProblemDocument2 pagesHow To Identify Maketing Research ProblempavanNo ratings yet
- Business Schools of AsiaDocument12 pagesBusiness Schools of AsiaRahul JainNo ratings yet
- SGX-Listed Hatten Land Diversifies Beyond Melaka Through Proposed Acquisition of Unicity in Seremban, MalaysiaDocument3 pagesSGX-Listed Hatten Land Diversifies Beyond Melaka Through Proposed Acquisition of Unicity in Seremban, MalaysiaWeR1 Consultants Pte LtdNo ratings yet
- Transmission Hydraulic SystemDocument4 pagesTransmission Hydraulic SystemA Ramos Gaby100% (5)
- AMM - 01-Aug-2019 - 79-21-10-000-004-A - Removal of The Lubrication UnitDocument10 pagesAMM - 01-Aug-2019 - 79-21-10-000-004-A - Removal of The Lubrication UnitIrfan05100% (1)