
Drugs and Cosmetics Act Part-II (BP-505T) (U-1, P-2)
Drugs and Cosmetics Act Part-II (BP-505T) (U-1, P-2)
You might also like
- AO 63 S 1989 (Read Sec 3.2)Document5 pagesAO 63 S 1989 (Read Sec 3.2)KarlaBadong14% (7)
- ABES IDC Estudo Mercado Brasileirode Software 2023 v02 PreviaDocument55 pagesABES IDC Estudo Mercado Brasileirode Software 2023 v02 PreviaDaniel SiesesNo ratings yet
- The Khyber Pakhtunkhwa Drugs Rules 1982 PDFDocument8 pagesThe Khyber Pakhtunkhwa Drugs Rules 1982 PDFmuhammad qasim100% (1)
- Guidane Documents - Export NOCDocument8 pagesGuidane Documents - Export NOCsudeepbNo ratings yet
- Moh-Uae Pharmacy Federal Law in English1Document23 pagesMoh-Uae Pharmacy Federal Law in English1Dr-Usman Khan100% (1)
- Who Certification SchemeDocument34 pagesWho Certification SchemeJnanankur BhowmikNo ratings yet
- GMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsFrom EverandGMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsRating: 5 out of 5 stars5/5 (2)
- Manufacture DetailedDocument29 pagesManufacture Detailedfreesubscriptionpurpose18No ratings yet
- India Drugs Cosmetic RulesDocument8 pagesIndia Drugs Cosmetic RulessalvaleuvenNo ratings yet
- Iv FluidDocument23 pagesIv FluidReaderNo ratings yet
- 2 Combined Unit I and Unit II Part 2 Provisions of The ActDocument45 pages2 Combined Unit I and Unit II Part 2 Provisions of The ActmavushieditzNo ratings yet
- 4-Drug & Cosmetic Act Rules 1945Document15 pages4-Drug & Cosmetic Act Rules 1945vara prasadNo ratings yet
- Balochistan Drugs RulesDocument5 pagesBalochistan Drugs RulesWàrìs Ràfìqùé ßàlòçhNo ratings yet
- Chapter 4 (Cosmetic Act)Document10 pagesChapter 4 (Cosmetic Act)ashmun nishaNo ratings yet
- Central Drugs Standard Control Organization: Guidance Document For Test LicenceDocument31 pagesCentral Drugs Standard Control Organization: Guidance Document For Test LicenceVikram UllalNo ratings yet
- Herblas and Biologicals GuidelinesDocument64 pagesHerblas and Biologicals GuidelinesManas DhariyaNo ratings yet
- Drugs InspectorDocument15 pagesDrugs InspectorThejomayiNo ratings yet
- Punjab Rules 1988Document5 pagesPunjab Rules 1988Ajma Line0% (1)
- Drug Law MCQsDocument4 pagesDrug Law MCQsAsadNo ratings yet
- Functions of Drug Branch Health DepartmentDocument8 pagesFunctions of Drug Branch Health Departmentgreatatiq007No ratings yet
- Procedure / Rules For Sale of DrugsDocument4 pagesProcedure / Rules For Sale of DrugsMoayed AmirNo ratings yet
- Bengal Drug RulesDocument98 pagesBengal Drug RulesShimul HalderNo ratings yet
- Unit 3 Notes DRADocument22 pagesUnit 3 Notes DRAOyshi RaoNo ratings yet
- KP Drug Rule 1982Document50 pagesKP Drug Rule 1982Zafran KhanNo ratings yet
- Sale of DrugsDocument28 pagesSale of Drugsvyshnosudha100% (2)
- CDSCO GuidanceForIndustryDocument181 pagesCDSCO GuidanceForIndustrydeepakmaramwarNo ratings yet
- Ilovepdf MergedDocument69 pagesIlovepdf MergedJagdeep SinghNo ratings yet
- WHO - Guidelines On The Implementation of The WHO Certification Scheme On The Quality of Pharmaceutical Products Moving in International CommerceDocument9 pagesWHO - Guidelines On The Implementation of The WHO Certification Scheme On The Quality of Pharmaceutical Products Moving in International CommerceDonny LoNo ratings yet
- Drug Rule 1945 PDFDocument104 pagesDrug Rule 1945 PDFKamal ShovonNo ratings yet
- Prepared By: Nidhi Patel Roll No: 117 College: Kbiper Department: Regulatory AffairsDocument30 pagesPrepared By: Nidhi Patel Roll No: 117 College: Kbiper Department: Regulatory AffairsNidhi PatelNo ratings yet
- Loan License 24E Application FormDocument6 pagesLoan License 24E Application FormprapannraghavNo ratings yet
- Form 5Document4 pagesForm 5Munir DayaniNo ratings yet
- Drug Registration Requirements in SudanDocument21 pagesDrug Registration Requirements in Sudanjai murugeshNo ratings yet
- Introduction To CDSCODocument11 pagesIntroduction To CDSCOaadrika negiNo ratings yet
- Contrect ManufactureDocument3 pagesContrect ManufacturebilalmasNo ratings yet
- Bureau Circular 5 S 1997Document39 pagesBureau Circular 5 S 1997mrk_rlndNo ratings yet
- Medical Cannabis in South AfricaDocument12 pagesMedical Cannabis in South AfricaRalphHiggo100% (1)
- CoppDocument21 pagesCoppSanjana ChoukseNo ratings yet
- AO 56 S 1989 LTODocument13 pagesAO 56 S 1989 LTOmikan2No ratings yet
- Decree No 7 Measures For The Supervision and Administration of Medical Device Production enDocument29 pagesDecree No 7 Measures For The Supervision and Administration of Medical Device Production enAnonymous ihBB7CNo ratings yet
- BE GuidelineDocument3 pagesBE GuidelinePrince PavanNo ratings yet
- CM Section 21 Application FormDocument11 pagesCM Section 21 Application FormMitchellNo ratings yet
- Amendment in Rule 20 A and Schedule H (Contract Manufacturing)Document7 pagesAmendment in Rule 20 A and Schedule H (Contract Manufacturing)Abdul Wahab KhanNo ratings yet
- Form 5-ADocument4 pagesForm 5-AAbdullahAbroNo ratings yet
- Unit2 Drug Acts and Its AmendmentsDocument29 pagesUnit2 Drug Acts and Its AmendmentsDinesh YadavNo ratings yet
- CDRR'S QPIRA Training Workshop On Drug Registration: Eliza G. SisonDocument35 pagesCDRR'S QPIRA Training Workshop On Drug Registration: Eliza G. SisonAidee SmithNo ratings yet
- CDRR'S QPIRA Training Workshop On Drug Registration: Eliza G. SisonDocument35 pagesCDRR'S QPIRA Training Workshop On Drug Registration: Eliza G. SisonAidee SmithNo ratings yet
- Untitled 7Document1 pageUntitled 7avi kaleNo ratings yet
- The Practice of Regulatory AffairsDocument42 pagesThe Practice of Regulatory AffairsCool AnnieNo ratings yet
- Grant of Drug and Cosmetic Manufacturing License in Own PremDocument3 pagesGrant of Drug and Cosmetic Manufacturing License in Own PremAbhilash NarayananNo ratings yet
- Compilation of Philippine Pharmacy LawDocument68 pagesCompilation of Philippine Pharmacy LawNoman AliNo ratings yet
- Loan LicenseDocument2 pagesLoan Licensejohnancy19No ratings yet
- RULE 291.131 Pharmacies Compounding Non-Sterile Preparations PDFDocument6 pagesRULE 291.131 Pharmacies Compounding Non-Sterile Preparations PDFCarl CrowNo ratings yet
- Balochistan Drugs Rules 1983Document8 pagesBalochistan Drugs Rules 1983Rashad Ishaq0% (1)
- Kmag18 - PHV and Psur-1Document3 pagesKmag18 - PHV and Psur-1dopamNo ratings yet
- The FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished PharmaceuticalsFrom EverandThe FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished PharmaceuticalsNo ratings yet
- Manual for the Implementation of Environmental, Health, and Safety Standards for the Control of Locusts: December 2021From EverandManual for the Implementation of Environmental, Health, and Safety Standards for the Control of Locusts: December 2021No ratings yet
- Nonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsFrom EverandNonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsWilliam J. BrockNo ratings yet
- Web D Worksheet 3Document19 pagesWeb D Worksheet 3aditya polNo ratings yet
- CAPITALGAINS 3rdsep PDFDocument202 pagesCAPITALGAINS 3rdsep PDFPhani Kumar SomarajupalliNo ratings yet
- 2017 Winter Model Answer PaperDocument21 pages2017 Winter Model Answer PaperjohnNo ratings yet
- Infrastructure Planning and Delivery:: Best Practice Case StudiesDocument88 pagesInfrastructure Planning and Delivery:: Best Practice Case StudiesAnurag KalvaNo ratings yet
- General Assembly Scholarship 2012-2013 ApplicationDocument7 pagesGeneral Assembly Scholarship 2012-2013 ApplicationjoesosnowskiNo ratings yet
- Propeller ShaftDocument8 pagesPropeller ShaftlomejorNo ratings yet
- 16 Submudline Receptical Data SheetDocument3 pages16 Submudline Receptical Data Sheetyehia sayedNo ratings yet
- CurriculaDocument6 pagesCurriculaAngel ValentinNo ratings yet
- Growing ApricotDocument9 pagesGrowing ApricotRajNo ratings yet
- Bachmann 2004Document194 pagesBachmann 2004weetjewatNo ratings yet
- Ac 145-9 CHG 1Document78 pagesAc 145-9 CHG 1Ricky RamirezNo ratings yet
- FEU HO1 Audit of Inventories 2017 PDFDocument4 pagesFEU HO1 Audit of Inventories 2017 PDFJoshuaNo ratings yet
- Practical Financial Management 7th Edition Lasher Solution ManualDocument27 pagesPractical Financial Management 7th Edition Lasher Solution Manualharold100% (23)
- 13 Electrical Pole's TrasverseDocument7 pages13 Electrical Pole's TrasverseReza PahlepiNo ratings yet
- Oracle Exambible 1z0-083 Sample Question 2020-Oct-08 by Hayden 71q VceDocument11 pagesOracle Exambible 1z0-083 Sample Question 2020-Oct-08 by Hayden 71q VcejowbrowNo ratings yet
- Vinayaka COLONYDocument1 pageVinayaka COLONY2451-18-737-020 NANNAPURAJU KARTHIKEYANo ratings yet
- Petitioners, vs. Pepsi Cola Products Phils., Inc. and Pepsico Inc., RespondentsDocument5 pagesPetitioners, vs. Pepsi Cola Products Phils., Inc. and Pepsico Inc., RespondentsAngelo GabrilloNo ratings yet
- Unit III-Leasing and Hire PurchaseDocument50 pagesUnit III-Leasing and Hire PurchaseAkashik GgNo ratings yet
- Registration of SocietyDocument6 pagesRegistration of SocietyDeepak AroraNo ratings yet
- Route 22 23 September 2023Document3 pagesRoute 22 23 September 2023api-523195650No ratings yet
- Ramana Orginal Ap37tb4145 Bajaj Allianz General Insurance CompanyDocument5 pagesRamana Orginal Ap37tb4145 Bajaj Allianz General Insurance Companysarath potnuriNo ratings yet
- PSBRC Module 3 - Media Relations PowerpointDocument58 pagesPSBRC Module 3 - Media Relations PowerpointGlen LunaNo ratings yet
- Astm f2164 PDFDocument5 pagesAstm f2164 PDFLuis J Villa Roel Bullon100% (1)
- TSE Dec 19 - WebDocument52 pagesTSE Dec 19 - Web伟雄No ratings yet
- Graficos de Bombas PVBasicsDocument45 pagesGraficos de Bombas PVBasicsLuis Panti EkNo ratings yet
- Practical Guide To Securing Work From Anywhere Using Microsoft 365 Business PremiumDocument28 pagesPractical Guide To Securing Work From Anywhere Using Microsoft 365 Business PremiumPedro Palmeira RochaNo ratings yet
- Peer Mentoring in A University Jazz Ensemble: by Andrew GoodrichDocument27 pagesPeer Mentoring in A University Jazz Ensemble: by Andrew GoodrichMatthew BelyeuNo ratings yet
- Jis G3101Document14 pagesJis G3101Hưng Ngô50% (2)
- ProductUploadTutorial RenderosityDocument4 pagesProductUploadTutorial RenderositySilvia SaavedraNo ratings yet

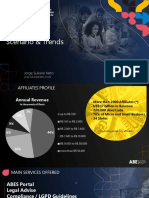























































































Download as pdf or txt
You might also like
- AO 63 S 1989 (Read Sec 3.2)Document5 pagesAO 63 S 1989 (Read Sec 3.2)KarlaBadong14% (7)
- ABES IDC Estudo Mercado Brasileirode Software 2023 v02 PreviaDocument55 pagesABES IDC Estudo Mercado Brasileirode Software 2023 v02 PreviaDaniel SiesesNo ratings yet
- The Khyber Pakhtunkhwa Drugs Rules 1982 PDFDocument8 pagesThe Khyber Pakhtunkhwa Drugs Rules 1982 PDFmuhammad qasim100% (1)
- Guidane Documents - Export NOCDocument8 pagesGuidane Documents - Export NOCsudeepbNo ratings yet
- Moh-Uae Pharmacy Federal Law in English1Document23 pagesMoh-Uae Pharmacy Federal Law in English1Dr-Usman Khan100% (1)
- Who Certification SchemeDocument34 pagesWho Certification SchemeJnanankur BhowmikNo ratings yet
- GMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsFrom EverandGMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsRating: 5 out of 5 stars5/5 (2)
- Manufacture DetailedDocument29 pagesManufacture Detailedfreesubscriptionpurpose18No ratings yet
- India Drugs Cosmetic RulesDocument8 pagesIndia Drugs Cosmetic RulessalvaleuvenNo ratings yet
- Iv FluidDocument23 pagesIv FluidReaderNo ratings yet
- 2 Combined Unit I and Unit II Part 2 Provisions of The ActDocument45 pages2 Combined Unit I and Unit II Part 2 Provisions of The ActmavushieditzNo ratings yet
- 4-Drug & Cosmetic Act Rules 1945Document15 pages4-Drug & Cosmetic Act Rules 1945vara prasadNo ratings yet
- Balochistan Drugs RulesDocument5 pagesBalochistan Drugs RulesWàrìs Ràfìqùé ßàlòçhNo ratings yet
- Chapter 4 (Cosmetic Act)Document10 pagesChapter 4 (Cosmetic Act)ashmun nishaNo ratings yet
- Central Drugs Standard Control Organization: Guidance Document For Test LicenceDocument31 pagesCentral Drugs Standard Control Organization: Guidance Document For Test LicenceVikram UllalNo ratings yet
- Herblas and Biologicals GuidelinesDocument64 pagesHerblas and Biologicals GuidelinesManas DhariyaNo ratings yet
- Drugs InspectorDocument15 pagesDrugs InspectorThejomayiNo ratings yet
- Punjab Rules 1988Document5 pagesPunjab Rules 1988Ajma Line0% (1)
- Drug Law MCQsDocument4 pagesDrug Law MCQsAsadNo ratings yet
- Functions of Drug Branch Health DepartmentDocument8 pagesFunctions of Drug Branch Health Departmentgreatatiq007No ratings yet
- Procedure / Rules For Sale of DrugsDocument4 pagesProcedure / Rules For Sale of DrugsMoayed AmirNo ratings yet
- Bengal Drug RulesDocument98 pagesBengal Drug RulesShimul HalderNo ratings yet
- Unit 3 Notes DRADocument22 pagesUnit 3 Notes DRAOyshi RaoNo ratings yet
- KP Drug Rule 1982Document50 pagesKP Drug Rule 1982Zafran KhanNo ratings yet
- Sale of DrugsDocument28 pagesSale of Drugsvyshnosudha100% (2)
- CDSCO GuidanceForIndustryDocument181 pagesCDSCO GuidanceForIndustrydeepakmaramwarNo ratings yet
- Ilovepdf MergedDocument69 pagesIlovepdf MergedJagdeep SinghNo ratings yet
- WHO - Guidelines On The Implementation of The WHO Certification Scheme On The Quality of Pharmaceutical Products Moving in International CommerceDocument9 pagesWHO - Guidelines On The Implementation of The WHO Certification Scheme On The Quality of Pharmaceutical Products Moving in International CommerceDonny LoNo ratings yet
- Drug Rule 1945 PDFDocument104 pagesDrug Rule 1945 PDFKamal ShovonNo ratings yet
- Prepared By: Nidhi Patel Roll No: 117 College: Kbiper Department: Regulatory AffairsDocument30 pagesPrepared By: Nidhi Patel Roll No: 117 College: Kbiper Department: Regulatory AffairsNidhi PatelNo ratings yet
- Loan License 24E Application FormDocument6 pagesLoan License 24E Application FormprapannraghavNo ratings yet
- Form 5Document4 pagesForm 5Munir DayaniNo ratings yet
- Drug Registration Requirements in SudanDocument21 pagesDrug Registration Requirements in Sudanjai murugeshNo ratings yet
- Introduction To CDSCODocument11 pagesIntroduction To CDSCOaadrika negiNo ratings yet
- Contrect ManufactureDocument3 pagesContrect ManufacturebilalmasNo ratings yet
- Bureau Circular 5 S 1997Document39 pagesBureau Circular 5 S 1997mrk_rlndNo ratings yet
- Medical Cannabis in South AfricaDocument12 pagesMedical Cannabis in South AfricaRalphHiggo100% (1)
- CoppDocument21 pagesCoppSanjana ChoukseNo ratings yet
- AO 56 S 1989 LTODocument13 pagesAO 56 S 1989 LTOmikan2No ratings yet
- Decree No 7 Measures For The Supervision and Administration of Medical Device Production enDocument29 pagesDecree No 7 Measures For The Supervision and Administration of Medical Device Production enAnonymous ihBB7CNo ratings yet
- BE GuidelineDocument3 pagesBE GuidelinePrince PavanNo ratings yet
- CM Section 21 Application FormDocument11 pagesCM Section 21 Application FormMitchellNo ratings yet
- Amendment in Rule 20 A and Schedule H (Contract Manufacturing)Document7 pagesAmendment in Rule 20 A and Schedule H (Contract Manufacturing)Abdul Wahab KhanNo ratings yet
- Form 5-ADocument4 pagesForm 5-AAbdullahAbroNo ratings yet
- Unit2 Drug Acts and Its AmendmentsDocument29 pagesUnit2 Drug Acts and Its AmendmentsDinesh YadavNo ratings yet
- CDRR'S QPIRA Training Workshop On Drug Registration: Eliza G. SisonDocument35 pagesCDRR'S QPIRA Training Workshop On Drug Registration: Eliza G. SisonAidee SmithNo ratings yet
- CDRR'S QPIRA Training Workshop On Drug Registration: Eliza G. SisonDocument35 pagesCDRR'S QPIRA Training Workshop On Drug Registration: Eliza G. SisonAidee SmithNo ratings yet
- Untitled 7Document1 pageUntitled 7avi kaleNo ratings yet
- The Practice of Regulatory AffairsDocument42 pagesThe Practice of Regulatory AffairsCool AnnieNo ratings yet
- Grant of Drug and Cosmetic Manufacturing License in Own PremDocument3 pagesGrant of Drug and Cosmetic Manufacturing License in Own PremAbhilash NarayananNo ratings yet
- Compilation of Philippine Pharmacy LawDocument68 pagesCompilation of Philippine Pharmacy LawNoman AliNo ratings yet
- Loan LicenseDocument2 pagesLoan Licensejohnancy19No ratings yet
- RULE 291.131 Pharmacies Compounding Non-Sterile Preparations PDFDocument6 pagesRULE 291.131 Pharmacies Compounding Non-Sterile Preparations PDFCarl CrowNo ratings yet
- Balochistan Drugs Rules 1983Document8 pagesBalochistan Drugs Rules 1983Rashad Ishaq0% (1)
- Kmag18 - PHV and Psur-1Document3 pagesKmag18 - PHV and Psur-1dopamNo ratings yet
- The FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished PharmaceuticalsFrom EverandThe FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished PharmaceuticalsNo ratings yet
- Manual for the Implementation of Environmental, Health, and Safety Standards for the Control of Locusts: December 2021From EverandManual for the Implementation of Environmental, Health, and Safety Standards for the Control of Locusts: December 2021No ratings yet
- Nonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsFrom EverandNonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsWilliam J. BrockNo ratings yet
- Web D Worksheet 3Document19 pagesWeb D Worksheet 3aditya polNo ratings yet
- CAPITALGAINS 3rdsep PDFDocument202 pagesCAPITALGAINS 3rdsep PDFPhani Kumar SomarajupalliNo ratings yet
- 2017 Winter Model Answer PaperDocument21 pages2017 Winter Model Answer PaperjohnNo ratings yet
- Infrastructure Planning and Delivery:: Best Practice Case StudiesDocument88 pagesInfrastructure Planning and Delivery:: Best Practice Case StudiesAnurag KalvaNo ratings yet
- General Assembly Scholarship 2012-2013 ApplicationDocument7 pagesGeneral Assembly Scholarship 2012-2013 ApplicationjoesosnowskiNo ratings yet
- Propeller ShaftDocument8 pagesPropeller ShaftlomejorNo ratings yet
- 16 Submudline Receptical Data SheetDocument3 pages16 Submudline Receptical Data Sheetyehia sayedNo ratings yet
- CurriculaDocument6 pagesCurriculaAngel ValentinNo ratings yet
- Growing ApricotDocument9 pagesGrowing ApricotRajNo ratings yet
- Bachmann 2004Document194 pagesBachmann 2004weetjewatNo ratings yet
- Ac 145-9 CHG 1Document78 pagesAc 145-9 CHG 1Ricky RamirezNo ratings yet
- FEU HO1 Audit of Inventories 2017 PDFDocument4 pagesFEU HO1 Audit of Inventories 2017 PDFJoshuaNo ratings yet
- Practical Financial Management 7th Edition Lasher Solution ManualDocument27 pagesPractical Financial Management 7th Edition Lasher Solution Manualharold100% (23)
- 13 Electrical Pole's TrasverseDocument7 pages13 Electrical Pole's TrasverseReza PahlepiNo ratings yet
- Oracle Exambible 1z0-083 Sample Question 2020-Oct-08 by Hayden 71q VceDocument11 pagesOracle Exambible 1z0-083 Sample Question 2020-Oct-08 by Hayden 71q VcejowbrowNo ratings yet
- Vinayaka COLONYDocument1 pageVinayaka COLONY2451-18-737-020 NANNAPURAJU KARTHIKEYANo ratings yet
- Petitioners, vs. Pepsi Cola Products Phils., Inc. and Pepsico Inc., RespondentsDocument5 pagesPetitioners, vs. Pepsi Cola Products Phils., Inc. and Pepsico Inc., RespondentsAngelo GabrilloNo ratings yet
- Unit III-Leasing and Hire PurchaseDocument50 pagesUnit III-Leasing and Hire PurchaseAkashik GgNo ratings yet
- Registration of SocietyDocument6 pagesRegistration of SocietyDeepak AroraNo ratings yet
- Route 22 23 September 2023Document3 pagesRoute 22 23 September 2023api-523195650No ratings yet
- Ramana Orginal Ap37tb4145 Bajaj Allianz General Insurance CompanyDocument5 pagesRamana Orginal Ap37tb4145 Bajaj Allianz General Insurance Companysarath potnuriNo ratings yet
- PSBRC Module 3 - Media Relations PowerpointDocument58 pagesPSBRC Module 3 - Media Relations PowerpointGlen LunaNo ratings yet
- Astm f2164 PDFDocument5 pagesAstm f2164 PDFLuis J Villa Roel Bullon100% (1)
- TSE Dec 19 - WebDocument52 pagesTSE Dec 19 - Web伟雄No ratings yet
- Graficos de Bombas PVBasicsDocument45 pagesGraficos de Bombas PVBasicsLuis Panti EkNo ratings yet
- Practical Guide To Securing Work From Anywhere Using Microsoft 365 Business PremiumDocument28 pagesPractical Guide To Securing Work From Anywhere Using Microsoft 365 Business PremiumPedro Palmeira RochaNo ratings yet
- Peer Mentoring in A University Jazz Ensemble: by Andrew GoodrichDocument27 pagesPeer Mentoring in A University Jazz Ensemble: by Andrew GoodrichMatthew BelyeuNo ratings yet
- Jis G3101Document14 pagesJis G3101Hưng Ngô50% (2)
- ProductUploadTutorial RenderosityDocument4 pagesProductUploadTutorial RenderositySilvia SaavedraNo ratings yet