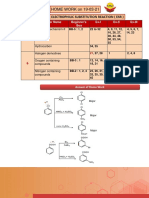
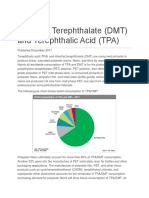
Download as pdf or txt
You might also like
- Organic Agriculture Production NC IiDocument36 pagesOrganic Agriculture Production NC IiJosephine Jane Echabarri80% (5)
- Kupchan Method of PartitioningDocument7 pagesKupchan Method of PartitioningRinta Moon100% (11)
- 304 Arndt Eistert Reaction PDFDocument8 pages304 Arndt Eistert Reaction PDFayesha sanaNo ratings yet
- Pinacol Pinacolone Rearrangement 1274Document14 pagesPinacol Pinacolone Rearrangement 1274Ayu RlNo ratings yet
- Wagner Meerwin Rearrangement: Reaction Conditions Mechanism Example Applications ProblemsDocument14 pagesWagner Meerwin Rearrangement: Reaction Conditions Mechanism Example Applications Problemsazeempharmacist4773No ratings yet
- 148 16SCCCH8 2020062412102615Document25 pages148 16SCCCH8 2020062412102615Jawad MughalNo ratings yet
- Pharmaceutical Organic Chemistry-I (BP202T) B. PHARM. FIRST YEAR (Second Sem.)Document18 pagesPharmaceutical Organic Chemistry-I (BP202T) B. PHARM. FIRST YEAR (Second Sem.)Kavi100% (1)
- 10 Carbonium Ion RearrangementsDocument56 pages10 Carbonium Ion Rearrangementssafiabarkat5No ratings yet
- Junaid and ImranDocument29 pagesJunaid and ImranJunaid KhanNo ratings yet
- 310 Beckmann RearrangementDocument7 pages310 Beckmann RearrangementHussain ShahidNo ratings yet
- 311 Pinacol Pinacolone RearrangementDocument9 pages311 Pinacol Pinacolone Rearrangementayesha sana100% (1)
- Molecular RearrangementsDocument40 pagesMolecular RearrangementsShravani SalunkheNo ratings yet
- Socl N Ag R-Cooh - R-Cocl - R-Cochn - R-CH Conu Ether, Et N Nucleophile (Nu)Document37 pagesSocl N Ag R-Cooh - R-Cocl - R-Cochn - R-CH Conu Ether, Et N Nucleophile (Nu)Shanmugam RameshNo ratings yet
- The Most Well-Known Rearrangements in Organic Chemistry at HandDocument32 pagesThe Most Well-Known Rearrangements in Organic Chemistry at HandAnkit JagetiaNo ratings yet
- Passing Package, Haloalkanes and HaloarenesDocument8 pagesPassing Package, Haloalkanes and HaloarenesShalem JeldiNo ratings yet
- Base HydrolysisDocument8 pagesBase Hydrolysissaud100% (1)
- Named ReactionDocument21 pagesNamed ReactionVijaya RajuNo ratings yet
- Poc Unit-5Document9 pagesPoc Unit-5Bintoo SharmaNo ratings yet
- RearrangementsDocument22 pagesRearrangementsVipul Newaskar100% (1)
- Reaction MechanismDocument19 pagesReaction Mechanismtapas kunduNo ratings yet
- RearrangementsDocument115 pagesRearrangementsscadvijayNo ratings yet
- Organic Chemistry-All Reaction MechanismsDocument30 pagesOrganic Chemistry-All Reaction MechanismsParameshwari kumar100% (1)
- Organic Aldehyde - Isothiocyanate ChemistryDocument244 pagesOrganic Aldehyde - Isothiocyanate Chemistrycarlosazucena1100% (2)
- Alkyne Zipper ReactionDocument28 pagesAlkyne Zipper ReactionRajeswari RajiNo ratings yet
- Organic ReactionsDocument44 pagesOrganic ReactionsJanhavi PatilNo ratings yet
- Types of Organic ReactionsDocument31 pagesTypes of Organic ReactionsNurulMAprilia80% (5)
- Catalytic Hydrogenations: Birch ReductionDocument9 pagesCatalytic Hydrogenations: Birch ReductionAnamikaNo ratings yet
- 1st Chap... Organic Reactions & Mechanisms111Document18 pages1st Chap... Organic Reactions & Mechanisms111Mrym NbNo ratings yet
- Deperoxidation of Cyclohexyl Hydroperoxide by Silica-Supported Alkoxo-Tantalum ComplexesDocument8 pagesDeperoxidation of Cyclohexyl Hydroperoxide by Silica-Supported Alkoxo-Tantalum ComplexesZhalaNo ratings yet
- Halogen Derivative of AlkaneDocument29 pagesHalogen Derivative of AlkaneDeepti Kaskar60% (5)
- 1995 Kinetic Aspects in The Selectivity of Deep HydrogenationDocument14 pages1995 Kinetic Aspects in The Selectivity of Deep Hydrogenationyusniya skNo ratings yet
- Pinacol Pinacolone Rearrangement QuestionsDocument7 pagesPinacol Pinacolone Rearrangement QuestionsM charan X CNo ratings yet
- Aldol CondensationDocument4 pagesAldol CondensationSreeja SatheeshNo ratings yet
- Phase TransferDocument8 pagesPhase Transfer0921pyNo ratings yet
- Aldol CondensationDocument71 pagesAldol CondensationJeyanthiNo ratings yet
- GssDocument26 pagesGssvnikhar123100% (1)
- Wolf Kishner ReactionDocument2 pagesWolf Kishner ReactionKonstantina Ms100% (1)
- SCYA7302Document95 pagesSCYA7302QueenNo ratings yet
- Mechanism of Organic ReactionsDocument45 pagesMechanism of Organic ReactionsBapu ThoratNo ratings yet
- MSC Chemistry Paper-III Unit-9Document24 pagesMSC Chemistry Paper-III Unit-9SIMARAN JAISWAL 41 M3SNo ratings yet
- Aknowledgement: "Photochemistry of Carbonyl Compounds" Which Helped Me To GainDocument24 pagesAknowledgement: "Photochemistry of Carbonyl Compounds" Which Helped Me To GainSmitaNo ratings yet
- Selenoxide EliminationDocument24 pagesSelenoxide EliminationKamal Kishor ThakurNo ratings yet
- Quinolines and IsoquinolinesDocument32 pagesQuinolines and IsoquinolinesPatel Vivek100% (1)
- Solvent-Free Microwave-Mediated Michael Addition ReactionsDocument6 pagesSolvent-Free Microwave-Mediated Michael Addition ReactionsDeepak sahooNo ratings yet
- Alkenes and AlkynesDocument22 pagesAlkenes and AlkynesAyodele AdeyonuNo ratings yet
- Experiment 12Document17 pagesExperiment 12Yvince LohNo ratings yet
- 1455785668CHE P9 M19 EtextDocument17 pages1455785668CHE P9 M19 EtextAuwal ShehuNo ratings yet
- 18.0 Carbonyl CompoundsDocument9 pages18.0 Carbonyl CompoundsKudzayi Tusaumwe100% (1)
- Submitted by Department of Chemistry, Imam Hossein University, Tehran, IRANDocument4 pagesSubmitted by Department of Chemistry, Imam Hossein University, Tehran, IRANsinaNo ratings yet
- Reductive Amination - WikipediaDocument13 pagesReductive Amination - Wikipediakhadar0001No ratings yet
- Portfolio in Orgchem FinalmanglicmotDocument8 pagesPortfolio in Orgchem FinalmanglicmotrlpmanglicmotNo ratings yet
- Subject ChemistryDocument13 pagesSubject Chemistryana pertiwiNo ratings yet
- Important MechanismsDocument11 pagesImportant Mechanismsyimisa2927No ratings yet
- Ch29 Brown Organic Chemistry SolutionsDocument8 pagesCh29 Brown Organic Chemistry SolutionsGabby Tanaka100% (1)
- Slide 1Document26 pagesSlide 1ShreyaNo ratings yet
- Benzilic Acid RearrangementDocument4 pagesBenzilic Acid Rearrangementachyuta.1433No ratings yet
- 1 Beckmann Rearrangement ReactionDocument6 pages1 Beckmann Rearrangement ReactionHifza BinyaminNo ratings yet
- Organic Reaction MechanismDocument32 pagesOrganic Reaction MechanismSaanvi MalhotraNo ratings yet
- Halogenalkanes: Unit 2 Chemistry C. Bailey PolackDocument23 pagesHalogenalkanes: Unit 2 Chemistry C. Bailey PolackBritney PattersonNo ratings yet
- Sustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeFrom EverandSustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeNo ratings yet
- Advances in Organometallic Chemistry and Catalysis: The Silver / Gold Jubilee International Conference on Organometallic Chemistry Celebratory BookFrom EverandAdvances in Organometallic Chemistry and Catalysis: The Silver / Gold Jubilee International Conference on Organometallic Chemistry Celebratory BookArmando J. L. PombeiroRating: 5 out of 5 stars5/5 (1)
- Chemical Tanker Construction, Systems and EquipmentDocument105 pagesChemical Tanker Construction, Systems and Equipmentandrej.sukoraNo ratings yet
- Post16organicsynthesisquizv1 322597Document19 pagesPost16organicsynthesisquizv1 322597diceplayaaNo ratings yet
- Phchem 1a - Activity #2 - Pharmaceutical Aids and NecessitiesDocument2 pagesPhchem 1a - Activity #2 - Pharmaceutical Aids and NecessitiesHercy AlintonNo ratings yet
- Leading Bio Pesticide, Bio Larvicide, Bío Fertilizer ManufacturerDocument36 pagesLeading Bio Pesticide, Bio Larvicide, Bío Fertilizer Manufacturervolkschem CropNo ratings yet
- Soil Fertility and Management3Document141 pagesSoil Fertility and Management3Pca ZdnNo ratings yet
- TextileTreatments e PDFDocument20 pagesTextileTreatments e PDFoverlord5555No ratings yet
- BCH 3120 Course Outline 2015Document3 pagesBCH 3120 Course Outline 2015Isuri VidyarathneNo ratings yet
- Design and Synthesis of Zinc (Ii) Complexes With Schiff Base Derived From 6-Aminopenicillanic Acid and Heterocyclic AldehydesDocument6 pagesDesign and Synthesis of Zinc (Ii) Complexes With Schiff Base Derived From 6-Aminopenicillanic Acid and Heterocyclic AldehydesIJAR JOURNALNo ratings yet
- Jecfa Co2 2006Document4 pagesJecfa Co2 2006Stella Lupita June TjandraNo ratings yet
- SDS - Rando HDZ 15Document11 pagesSDS - Rando HDZ 15Georgina SuleNo ratings yet
- Genetic Analysis An Integrated Approach 2nd Edition Sanders Test Bank 1Document14 pagesGenetic Analysis An Integrated Approach 2nd Edition Sanders Test Bank 1theresa100% (43)
- Scott 2001Document20 pagesScott 2001Mariana CatiniNo ratings yet
- 3.6.5. One-Shot AcidDocument1 page3.6.5. One-Shot Acidpedro taquichiriNo ratings yet
- Refinery BasicsDocument27 pagesRefinery BasicsArsalan QadirNo ratings yet
- Amantadine HCL CapsulesDocument2 pagesAmantadine HCL CapsulesDinie NoviantyNo ratings yet
- 1.mechanism of PolymerizationDocument6 pages1.mechanism of PolymerizationShalynee SuthaharNo ratings yet
- Caprylic AcidDocument8 pagesCaprylic AcidVamsi KrishnaNo ratings yet
- Solutions Chromatography 210111 Examination TFKE30Document3 pagesSolutions Chromatography 210111 Examination TFKE30MislavNo ratings yet
- Answer All Questions in This SectionDocument6 pagesAnswer All Questions in This SectionAri AdiantariNo ratings yet
- Lecture 16Document15 pagesLecture 16Fabiha Shafi MimNo ratings yet
- AdditivsDocument40 pagesAdditivsMohsin MalikNo ratings yet
- Urea-Formaldehyde Adhesive Resins: Anthony H. ConnerDocument8 pagesUrea-Formaldehyde Adhesive Resins: Anthony H. ConnerAditiya Muchsin SobariNo ratings yet
- Kebutuhan DMT AsiaDocument2 pagesKebutuhan DMT Asiafebry16pwjNo ratings yet
- 4.2.2 Polyesters and PolyamidesDocument7 pages4.2.2 Polyesters and PolyamidesttsNo ratings yet
- OGA - Chemical Series - Soda Ash Market Outlook 2019-2025Document20 pagesOGA - Chemical Series - Soda Ash Market Outlook 2019-2025ambarish ramNo ratings yet
- Vet McqsDocument133 pagesVet McqsNiaz Asghar63% (8)
- Continuous Ethanol Fermentation Using Immobilized YeastDocument1 pageContinuous Ethanol Fermentation Using Immobilized YeastAlejoldmNo ratings yet
- List of On-Roll Ph.D. Scholars at Thapar University, Patiala As On 29.06.2017Document18 pagesList of On-Roll Ph.D. Scholars at Thapar University, Patiala As On 29.06.2017SumanNo ratings yet