Download as key, pdf, or txt
You might also like
- Ebook PDF Developing Content Area Literacy 40 Strategies For Middle and Secondary Classrooms 2nd Edition PDFDocument41 pagesEbook PDF Developing Content Area Literacy 40 Strategies For Middle and Secondary Classrooms 2nd Edition PDFviolet.campos327100% (45)
- The Covid Vaccine Linked To CancerDocument34 pagesThe Covid Vaccine Linked To CancerNoyb Nal100% (4)
- MODULE 1 Electric PotentialDocument10 pagesMODULE 1 Electric Potentialjovy dulayNo ratings yet
- Prebiotic Systems Chemistry - New Perspectives For The Origins of LifeDocument82 pagesPrebiotic Systems Chemistry - New Perspectives For The Origins of LifeMartin WolframNo ratings yet
- Cheddar - RDocument6 pagesCheddar - Rginebra rivera lazarteNo ratings yet
- Research Statement: Saket Navlakha School of Computer Science Carnegie Mellon University, Pittsburgh, PADocument4 pagesResearch Statement: Saket Navlakha School of Computer Science Carnegie Mellon University, Pittsburgh, PAARK ZIFARNo ratings yet
- Mass Spectrometry-Based Glycoprofiling of Biopharmaceuticals by Using An Automated Data Processing Tool - SimGlycan®Document10 pagesMass Spectrometry-Based Glycoprofiling of Biopharmaceuticals by Using An Automated Data Processing Tool - SimGlycan®yun baiNo ratings yet
- Fast and Sensitive Taxonomic Assignment To Metagenomic ContigsDocument3 pagesFast and Sensitive Taxonomic Assignment To Metagenomic Contigsariel_jNo ratings yet
- 498 Prado et al. - Urochloa and Maize rotationDocument1 page498 Prado et al. - Urochloa and Maize rotationJyonNo ratings yet
- Borges 2021 QNova MzMIneDocument14 pagesBorges 2021 QNova MzMIneRodolpho GuilhermeNo ratings yet
- BPSM 4,5Document25 pagesBPSM 4,5Aditya KshirsagarNo ratings yet
- Joanes Grandjean A Consensus Protocol For FunctionalDocument25 pagesJoanes Grandjean A Consensus Protocol For Functionalvladce.trNo ratings yet
- MINION Data AnalisiDocument10 pagesMINION Data AnalisijacorvarNo ratings yet
- Metaboanalyst 4.0: Towards More Transparent and Integrative Metabolomics AnalysisDocument10 pagesMetaboanalyst 4.0: Towards More Transparent and Integrative Metabolomics AnalysisNi Putu Ayu Erninda Oktaviani SuputriNo ratings yet
- M.SC DA Syllabus 2017 19 BatchDocument64 pagesM.SC DA Syllabus 2017 19 BatchMadhumithaNo ratings yet
- Carotenoid and Tocopherol Fortification of Zucchini Fruits Using A Viral RNA VectorDocument1 pageCarotenoid and Tocopherol Fortification of Zucchini Fruits Using A Viral RNA VectorOmkar MudkannaNo ratings yet
- Chaitanya Bharathi Institute of Technology Department of Biotechnology B.Tech IV - Year Semester - IDocument25 pagesChaitanya Bharathi Institute of Technology Department of Biotechnology B.Tech IV - Year Semester - IRajashekhar BogarajulaNo ratings yet
- Recursive Feature Elimination WithDocument9 pagesRecursive Feature Elimination WithYakin BagusNo ratings yet
- 18BTB103T - Human Physiology and Health SyllabusDocument2 pages18BTB103T - Human Physiology and Health SyllabusSounak NandyNo ratings yet
- 2018 Luque de CastroDocument9 pages2018 Luque de CastroKeyla MaydeeNo ratings yet
- Amezquita Et Al - 2020 - Orchestrating Single-Cell Analysis With BioconductorDocument9 pagesAmezquita Et Al - 2020 - Orchestrating Single-Cell Analysis With Bioconductorkeon.arbabi.altNo ratings yet
- Deep Learning Driven Biosynthetic Pathways Navigation For Natural Products With Bionavi-NpDocument9 pagesDeep Learning Driven Biosynthetic Pathways Navigation For Natural Products With Bionavi-NpElias Gerardo Pardo LlamoccaNo ratings yet
- F1000research 273611Document1 pageF1000research 273611azjajaoan malayaNo ratings yet
- MorphoTools2 PaperDocument2 pagesMorphoTools2 Paperdeneb denebNo ratings yet
- PRACTICAL RESEARCH 1 (Module1-2) : Bonifacio R. Tagaban Sr. Integrated SchoolDocument1 pagePRACTICAL RESEARCH 1 (Module1-2) : Bonifacio R. Tagaban Sr. Integrated SchoolLoraine DiazonNo ratings yet
- Project Report - Validation of Time Seri PDFDocument31 pagesProject Report - Validation of Time Seri PDFAmmr MahmoodNo ratings yet
- Ismb99 008Document8 pagesIsmb99 008Orion OriNo ratings yet
- Sri Astrology PanchagamDocument185 pagesSri Astrology PanchagamBharathwaj KomarrajuNo ratings yet
- Pharm Research ProteomicsDocument4 pagesPharm Research Proteomicsmsoile6No ratings yet
- The Best Approach For Functional Tricuspid Regurgitation. A Network Meta-AnalysisDocument13 pagesThe Best Approach For Functional Tricuspid Regurgitation. A Network Meta-AnalysisMixalisKaplanisNo ratings yet
- Molecular Systems Biology - 2019 - Luecken - Current Best Practices in Single Cell RNA Seq Analysis A TutorialDocument23 pagesMolecular Systems Biology - 2019 - Luecken - Current Best Practices in Single Cell RNA Seq Analysis A TutorialAditya ChakravartyNo ratings yet
- Vaccinegate:: Final Technical Report Molecular Profile Analysis of VaccineDocument4 pagesVaccinegate:: Final Technical Report Molecular Profile Analysis of VaccineYiota KokkorisNo ratings yet
- JNS AgriBiotech Vol 15 5Document9 pagesJNS AgriBiotech Vol 15 5HAJAR ZZNo ratings yet
- Europharmreview NIRDocument7 pagesEuropharmreview NIRMaria IshfaqNo ratings yet
- Development of An Animal-Component Free Electroporation and Recovery Formulation Using EX-CELL CHO Cloning MediumDocument1 pageDevelopment of An Animal-Component Free Electroporation and Recovery Formulation Using EX-CELL CHO Cloning MediumSAFC-GlobalNo ratings yet
- Massspectrometry 5 1 A0043Document6 pagesMassspectrometry 5 1 A0043Fadli SukandiarsyahNo ratings yet
- CoVVSURF Package - GeneurDocument23 pagesCoVVSURF Package - GeneurbiguncleNo ratings yet
- Thermal Methods of AnalysisDocument76 pagesThermal Methods of AnalysisAzuriak1100% (2)
- Sqmtools: Automated Processing and Visual Analysis of 'Omics Data With R and Anvi'ODocument11 pagesSqmtools: Automated Processing and Visual Analysis of 'Omics Data With R and Anvi'OshanmugapriyaNo ratings yet
- Lab Sheet All Force On A Sluice GateDocument9 pagesLab Sheet All Force On A Sluice GateMastura Binti HaronNo ratings yet
- Genetics and CytogeneticsDocument2 pagesGenetics and CytogeneticsSounak NandyNo ratings yet
- Learning Resource Pack Approval Sheet: Arlac Gricultural NiversityDocument5 pagesLearning Resource Pack Approval Sheet: Arlac Gricultural NiversityJohn Christian MejiaNo ratings yet
- 6220 15575 1 SM 1 Chtita PDFDocument8 pages6220 15575 1 SM 1 Chtita PDFVarun AwasthiNo ratings yet
- Towards Sustainable Roads Through High RAP Content LayersDocument1 pageTowards Sustainable Roads Through High RAP Content LayersAndréNagalliNo ratings yet
- Sirah ReviewDocument12 pagesSirah Reviewsoumava palitNo ratings yet
- RAFT-Mediated Polymerization-InducedSelf-AssemblyDocument25 pagesRAFT-Mediated Polymerization-InducedSelf-AssemblyRohit ChandolaNo ratings yet
- Mathematical Modeling of Gene Expression: A Guide For The Perplexed BiologistDocument15 pagesMathematical Modeling of Gene Expression: A Guide For The Perplexed Biologistcarlos ArozamenaNo ratings yet
- Gujarat Technological University: W.E.F. AY 2018-19Document4 pagesGujarat Technological University: W.E.F. AY 2018-19Shivam PanchalNo ratings yet
- Costa-Moura2020 Chapter ABibliometricAnalysisOfTheStraDocument15 pagesCosta-Moura2020 Chapter ABibliometricAnalysisOfTheStraRobson CostaNo ratings yet
- Bio 01 PDFDocument53 pagesBio 01 PDFmrcopy xeroxNo ratings yet
- Debinfer: Bayesian Inference For Dynamical Models of Biological Systems in RDocument27 pagesDebinfer: Bayesian Inference For Dynamical Models of Biological Systems in RshidaoNo ratings yet
- ChemoPower Pub - Coelute and Trance Components FindingDocument14 pagesChemoPower Pub - Coelute and Trance Components FindingShinichiHamanoNo ratings yet
- Use of Principal Component Analysis (PCA) and Hierarchical Cluster Analysis 2017Document8 pagesUse of Principal Component Analysis (PCA) and Hierarchical Cluster Analysis 2017Gustavo Andres Fuentes RodriguezNo ratings yet
- TaxonDocument200 pagesTaxonSimion MehedintiNo ratings yet
- BTY 405 IGAP FormatDocument4 pagesBTY 405 IGAP FormatShahil AlamNo ratings yet
- 2019SUMS-RAS artMS Jimenez-MoralesDocument31 pages2019SUMS-RAS artMS Jimenez-MoralesMayank GuptaNo ratings yet
- STP ParametersDocument8 pagesSTP Parametersakhilt29No ratings yet
- 18BTC106J-Immunology SyllabusDocument2 pages18BTC106J-Immunology SyllabusChris PenielNo ratings yet
- Course Curriculum PG 2023-2024 - 0Document50 pagesCourse Curriculum PG 2023-2024 - 0shyamNo ratings yet
- Pine CoineDocument18 pagesPine Coineahmad alzoubiNo ratings yet
- Mixture Models and ApplicationsFrom EverandMixture Models and ApplicationsNizar BouguilaNo ratings yet
- Purposive Communication (Part 1) Purposive Communication (Part 1)Document14 pagesPurposive Communication (Part 1) Purposive Communication (Part 1)Tricia Mae Comia AtienzaNo ratings yet
- Failure Modes of Rocks Under Uniaxial Compression Tests - HBRP PublicationDocument8 pagesFailure Modes of Rocks Under Uniaxial Compression Tests - HBRP PublicationVijay KNo ratings yet
- Class 7 Ch-4 Heat Notebook WorkDocument5 pagesClass 7 Ch-4 Heat Notebook WorkKPOP WORLDNo ratings yet
- College of Business and AccountancyDocument5 pagesCollege of Business and AccountancyJerico ManaloNo ratings yet
- The Fourth RayDocument11 pagesThe Fourth RayQizhi NeoNo ratings yet
- An Archaeological Investigation of Sira Bay, Aden, Republic of YemenDocument11 pagesAn Archaeological Investigation of Sira Bay, Aden, Republic of YemenAbdussalam NagiNo ratings yet
- 2nd Grade Journey's Unit 1 Week3 Day 1 Animal TraitsDocument49 pages2nd Grade Journey's Unit 1 Week3 Day 1 Animal TraitsDalia ElgamalNo ratings yet
- ExamEssentials Test 1Document4 pagesExamEssentials Test 1Ngân Hoàng Ngọc ThảoNo ratings yet
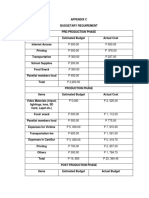
- Other AppendicesDocument9 pagesOther Appendicesbeverly bombitaNo ratings yet
- DS - ArraysDocument67 pagesDS - Arrayskalash satypalNo ratings yet
- Interaction Between Parallel Underground Openings: H. GerçekDocument9 pagesInteraction Between Parallel Underground Openings: H. GerçekEDWIN MEJIA MANRIQUENo ratings yet
- Newton'S Second Law: Driving Question - ObjectiveDocument6 pagesNewton'S Second Law: Driving Question - ObjectiveRKNo ratings yet
- Sound Absorption Prediction Software: Absorber Is A Computer Program ForDocument1 pageSound Absorption Prediction Software: Absorber Is A Computer Program Forchristian atauje garciaNo ratings yet
- DetailaDocument5 pagesDetailaNoneta MotsiNo ratings yet
- Plastic Surgery Persuasive EssayDocument4 pagesPlastic Surgery Persuasive Essayezm8kqbt100% (2)
- Curriculum - Chemist - Production (Pharma, Cosmetics - Biologics)Document41 pagesCurriculum - Chemist - Production (Pharma, Cosmetics - Biologics)srinivas6321No ratings yet
- Circuits MCQ DR HaithamDocument7 pagesCircuits MCQ DR HaithamΜΔHΜΘƱD ЅHΓΣΣF.No ratings yet
- Logos (Heraclitus, Fragment B 50) : Ouk E Uou DXXD Tou Adyou Dkouoavraq OocpdvDocument20 pagesLogos (Heraclitus, Fragment B 50) : Ouk E Uou DXXD Tou Adyou Dkouoavraq OocpdvdugNo ratings yet
- Cooling Tower EvaporationDocument2 pagesCooling Tower Evaporationamg007No ratings yet
- 50-Increase & Decrease of RatioDocument6 pages50-Increase & Decrease of RatioSameh Salah100% (1)
- Safety Induction Visitor EngDocument16 pagesSafety Induction Visitor EngNico George100% (1)
- The TideDocument43 pagesThe Tideghaniya.juma04No ratings yet
- Transiluminador BenchtopDocument11 pagesTransiluminador BenchtopWendy Lilian Gómez MonsivaisNo ratings yet
- Elt Systems Service Information Letter: Date: May 6, 2020 Number: SIL4013Document4 pagesElt Systems Service Information Letter: Date: May 6, 2020 Number: SIL4013Nitesh PalNo ratings yet
- Cone Crusher Sell Sheet FinalDocument2 pagesCone Crusher Sell Sheet FinalVladimir Illich PinzonNo ratings yet
- Testing The HypothesisDocument2 pagesTesting The HypothesisJaqueline Mae AmaquinNo ratings yet
- MPT Part I & II Neurological Physiotherapy - PDF - Peripheral Neuropathy - Physical TherapyDocument36 pagesMPT Part I & II Neurological Physiotherapy - PDF - Peripheral Neuropathy - Physical TherapyYawer Hamaid LoneNo ratings yet
- Rotordynamic Analysis of The AM600 Turbine-Generator ShaftlineDocument20 pagesRotordynamic Analysis of The AM600 Turbine-Generator ShaftlineJarot PrakosoNo ratings yet