
Geneticsdiseases 1
Geneticsdiseases 1
You might also like
- The RMBC MethodDocument128 pagesThe RMBC MethodFaizan Ahmed100% (2)
- Fce 2015 Complete Tests 1 e 2 AnswersDocument4 pagesFce 2015 Complete Tests 1 e 2 AnswersMárcia Bonfim83% (23)
- 21-Day Mind, Body and Spirit DetoxDocument6 pages21-Day Mind, Body and Spirit DetoxAjay TiwariNo ratings yet
- Cystic Fibrosis: Zoe Drullard and Avril GuillermoDocument18 pagesCystic Fibrosis: Zoe Drullard and Avril Guillermozoe drullardNo ratings yet
- Cystic Fibrosis: Gene TherapyDocument19 pagesCystic Fibrosis: Gene TherapyChansiekeiNo ratings yet
- Cystic FibrosisDocument3 pagesCystic Fibrosissumana yeasminNo ratings yet
- Cystic FibrosisDocument35 pagesCystic Fibrosisvani reddyNo ratings yet
- Cystic FibrosisDocument6 pagesCystic Fibrosischurchil owinoNo ratings yet
- The Cell Biology of Cystic FibrosisDocument23 pagesThe Cell Biology of Cystic FibrosisShamarie Love MaribaoNo ratings yet
- Theme1AppliedLecture2 BIO1A03Fall2022Document27 pagesTheme1AppliedLecture2 BIO1A03Fall2022proplayer910No ratings yet
- Cystic FibrosisDocument3 pagesCystic FibrosisstanscimagNo ratings yet
- Biochemistry Presentation.Document3 pagesBiochemistry Presentation.MIMIbako67No ratings yet
- Cystic FibrosisDocument14 pagesCystic FibrosisAishistic thingsNo ratings yet
- 16.5 Gene Therapy: 10.1 CoordinationDocument18 pages16.5 Gene Therapy: 10.1 Coordinationsohailnoreen5062No ratings yet
- Cystic Fibrosis: Bio FactsheetDocument3 pagesCystic Fibrosis: Bio FactsheetJavier SaadNo ratings yet
- Cystic Fibrosis: Dr. Pragasam Viswanathan Professor, SBSTDocument16 pagesCystic Fibrosis: Dr. Pragasam Viswanathan Professor, SBSTMaru Mengesha Worku 18BBT0285No ratings yet
- 1.3.7 Cystic Fibrosis: Chapter 3 Cystic Fibrosis HBIO1 - The Body and Its Diseases AQA AS Human BiologyDocument22 pages1.3.7 Cystic Fibrosis: Chapter 3 Cystic Fibrosis HBIO1 - The Body and Its Diseases AQA AS Human BiologyMaha NaserNo ratings yet
- The Genetics of Cystic FibrosisDocument24 pagesThe Genetics of Cystic FibrosisThrilok Nath MadduruNo ratings yet
- Cysticfibrosisdonebymyfriend 120218095057 Phpapp02Document28 pagesCysticfibrosisdonebymyfriend 120218095057 Phpapp02Elham AlwagaaNo ratings yet
- Cystic FibrosisDocument21 pagesCystic FibrosisGouthamNo ratings yet
- J-2.16 Bio1201Document7 pagesJ-2.16 Bio1201gert16No ratings yet
- CR Project On Cystic FibrosisDocument3 pagesCR Project On Cystic FibrosisMorgan PeggNo ratings yet
- Cystic FibrosisDocument9 pagesCystic FibrosiszarasarNo ratings yet
- Genes and Patterns of InheritanceDocument25 pagesGenes and Patterns of InheritanceNeelam HanifNo ratings yet
- Anaphy-Case Study-CjbascoDocument3 pagesAnaphy-Case Study-CjbascoChristine Jane BascoNo ratings yet
- Cystic Fibrosis Fact SheetDocument6 pagesCystic Fibrosis Fact SheetAnggun SafariantiniNo ratings yet
- Microsoft PowerPoint Presentation جديدDocument10 pagesMicrosoft PowerPoint Presentation جديدakram alrdayNo ratings yet
- Cystic Fibrosis With Case StudyDocument27 pagesCystic Fibrosis With Case StudyAviva Tuteja100% (1)
- ITW - Cystic Fibrosis Lecture 1Document9 pagesITW - Cystic Fibrosis Lecture 1Maha KhanNo ratings yet
- Cystic Fibrosis (CF) Final 1Document34 pagesCystic Fibrosis (CF) Final 1Mapis BravoNo ratings yet
- Cystic FibrosisDocument29 pagesCystic FibrosisAviva TutejaNo ratings yet
- Cystic FibrosisDocument22 pagesCystic FibrosisIeva PetrylaiteNo ratings yet
- Cystic FibrosisDocument19 pagesCystic FibrosisChristian Dene Opada TatoyNo ratings yet
- Genetic Disease ChartDocument1 pageGenetic Disease ChartNellena Kaye PedrigalNo ratings yet
- Case Study: Cystic Fibrosis: Manalo, Joanne Erica S. BS Respiratory TherapyDocument20 pagesCase Study: Cystic Fibrosis: Manalo, Joanne Erica S. BS Respiratory TherapyJohn Carlo ConsultaNo ratings yet
- A New Hope. Trikafta For The Treatment of Cystic FibrosisDocument6 pagesA New Hope. Trikafta For The Treatment of Cystic FibrosisTania CarreñoNo ratings yet
- CFTR GeneticsreviewDocument12 pagesCFTR GeneticsreviewTanya GreyNo ratings yet
- Diseases of Pattern of InheritanceDocument7 pagesDiseases of Pattern of InheritanceLABORATORY ASTMMCNo ratings yet
- Cystic FibrosisDocument23 pagesCystic FibrosisawirbshtiwanNo ratings yet
- Cystic FibrosisDocument71 pagesCystic Fibrosishumera0% (1)
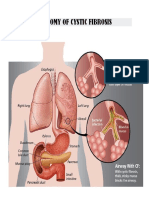
- Anatomy of Cystic FibrosisDocument5 pagesAnatomy of Cystic FibrosislazylawatudentNo ratings yet
- Cystic Fibrosis: Nature Reviews Disease Primers May 2015Document20 pagesCystic Fibrosis: Nature Reviews Disease Primers May 2015Tapan AnkleshwariaNo ratings yet
- Cystic Fibrosis Pancreas: Deevon M. Cariaga FEU-NRMF MedicineDocument7 pagesCystic Fibrosis Pancreas: Deevon M. Cariaga FEU-NRMF MedicinedeevoncNo ratings yet
- Cystic FibrosisDocument13 pagesCystic FibrosisAnkita guptaNo ratings yet
- Ewence2020Cystic FibrosisDocument5 pagesEwence2020Cystic FibrosismacedovendezuNo ratings yet
- A Review of Trikafta: Triple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulator TherapyDocument7 pagesA Review of Trikafta: Triple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulator TherapySugi AntoNo ratings yet
- Cystic FibrosisDocument16 pagesCystic FibrosisAzzahra AzmiNo ratings yet
- Cystic FibrosisDocument33 pagesCystic FibrosisGreeshma ShajiNo ratings yet
- Cystic Fibrosis Research NotesDocument8 pagesCystic Fibrosis Research NotesNicole KrawitzNo ratings yet
- Cystic Fibrosis Case ReportDocument17 pagesCystic Fibrosis Case ReportMarjo100% (1)
- Columnaris: Manimozhi. E AAH-MA8-05 Aahm, CifeDocument30 pagesColumnaris: Manimozhi. E AAH-MA8-05 Aahm, CifeManimozhi EthirajanNo ratings yet
- Fibrosis Quística - NEJM02112023Document15 pagesFibrosis Quística - NEJM02112023Jesus A. Pineda GarciaNo ratings yet
- Cystic FibrosisDocument20 pagesCystic FibrosisIndrajit BaruaNo ratings yet
- Pemicu 5 Cessy ChristyDocument205 pagesPemicu 5 Cessy Christygrace liwantoNo ratings yet
- Cystic FibrosisDocument20 pagesCystic FibrosisIndrajit Barua100% (1)
- Cystic FibrosisDocument2 pagesCystic Fibrosisthatkid94No ratings yet
- Review of Rapid Advances in Cystic FibrosisDocument7 pagesReview of Rapid Advances in Cystic Fibrosismihaelaatanasova123No ratings yet
- Cystic FibrosisDocument32 pagesCystic FibrosisSrm Genetics100% (1)
- Cystic FibrosisDocument40 pagesCystic FibrosisMoonmoon SinhaNo ratings yet
- Adaptation To High Rates of Chromosomal Instability and Aneuploidy Through Multiple Pathways in Budding YeastDocument19 pagesAdaptation To High Rates of Chromosomal Instability and Aneuploidy Through Multiple Pathways in Budding YeastFernandoNo ratings yet
- Cysticfibrosis PDFDocument20 pagesCysticfibrosis PDFTORRES Hazel Ann I.No ratings yet
- CysticfibrosisDocument6 pagesCysticfibrosisSghir SghirNo ratings yet
- Organisational BehaviourDocument279 pagesOrganisational BehaviourSumitha SelvarajNo ratings yet
- The Literary Comparison Contrast EssayDocument7 pagesThe Literary Comparison Contrast EssayThapelo SebolaiNo ratings yet
- Conceptual Design of An Automotive Composite Brake Pedal: Mohd Sapuan Salit, Mohd Syed Ali Molla and MD Liakot AliDocument5 pagesConceptual Design of An Automotive Composite Brake Pedal: Mohd Sapuan Salit, Mohd Syed Ali Molla and MD Liakot Aliseran ünalNo ratings yet
- 2011 Diving Catalogue BEUCHATDocument60 pages2011 Diving Catalogue BEUCHATRalph KramdenNo ratings yet
- Third Party Logistics: A Literature Review and Research AgendaDocument26 pagesThird Party Logistics: A Literature Review and Research AgendaRuxandra Radus100% (1)
- Justin Bieber Is - Famous Singer.: (You Must Read Out The Whole Sentence.)Document2 pagesJustin Bieber Is - Famous Singer.: (You Must Read Out The Whole Sentence.)Irene De la FuenteNo ratings yet
- Cosmetics Case ProjectDocument16 pagesCosmetics Case ProjectLaura NardiNo ratings yet
- Reactor and Regenerator System of FCCDocument67 pagesReactor and Regenerator System of FCCDai NamNo ratings yet
- The DJ Test: Personalised Report and Recommendations For Alex YachevskiDocument34 pagesThe DJ Test: Personalised Report and Recommendations For Alex YachevskiSashadanceNo ratings yet
- CASHe Soa 568304-1Document2 pagesCASHe Soa 568304-1sathyaNo ratings yet
- Lab 6 Shouq PDFDocument10 pagesLab 6 Shouq PDFreemy100% (1)
- BSI BCI Horizon Scan Report 2016 PDFDocument32 pagesBSI BCI Horizon Scan Report 2016 PDFSandraNo ratings yet
- Small Unit Operations: PMA Core Values: Selfless Service To God and Country, Honor, ExcellenceDocument33 pagesSmall Unit Operations: PMA Core Values: Selfless Service To God and Country, Honor, ExcellenceAbay GeeNo ratings yet
- Full Download Art of Leadership 5th Edition Manning Test Bank PDF Full ChapterDocument36 pagesFull Download Art of Leadership 5th Edition Manning Test Bank PDF Full Chaptermasqueedenized8a43l100% (19)
- What Do You Know About Jobs - 28056Document2 pagesWhat Do You Know About Jobs - 28056Kadek DharmawanNo ratings yet
- Accounting Standards - Handwritten Notes - Udesh Regular - Group 1Document24 pagesAccounting Standards - Handwritten Notes - Udesh Regular - Group 1Uday TomarNo ratings yet
- Past Progressive SeptimoDocument1 pagePast Progressive Septimoferney cordobaNo ratings yet
- Bsl6 Qual Spec 11-12 FinalDocument35 pagesBsl6 Qual Spec 11-12 FinalBSLcourses.co.ukNo ratings yet
- Postmodern Nihilism Theory and LiteratureDocument336 pagesPostmodern Nihilism Theory and LiteratureCátia Tavares100% (3)
- Comparative and Superlative Adjectives PresentationDocument15 pagesComparative and Superlative Adjectives Presentationapi-248688582No ratings yet
- Code of Ethics For Professional TeachersDocument12 pagesCode of Ethics For Professional TeachersJean GuyuranNo ratings yet
- GST/HST Credit Application For Individuals Who Become Residents of CanadaDocument4 pagesGST/HST Credit Application For Individuals Who Become Residents of CanadaAndrea Dr FanisNo ratings yet
- PCA Churches in CaliforniaDocument4 pagesPCA Churches in CaliforniaLIGHT GNo ratings yet
- Deftones - Back To School (Pink Maggit Rap Version) : (Intro)Document2 pagesDeftones - Back To School (Pink Maggit Rap Version) : (Intro)zwartwerkerijNo ratings yet
- Effect of Bolt Pretension in Single Lap Bolted Joint IJERTV4IS010269Document4 pagesEffect of Bolt Pretension in Single Lap Bolted Joint IJERTV4IS010269ayush100% (1)
- Womens EssentialsDocument50 pagesWomens EssentialsNoor-uz-Zamaan AcademyNo ratings yet
- Petitioner vs. VS.: Third DivisionDocument12 pagesPetitioner vs. VS.: Third DivisionAnonymous QR87KCVteNo ratings yet
























































































Download as pdf or txt
You might also like
- The RMBC MethodDocument128 pagesThe RMBC MethodFaizan Ahmed100% (2)
- Fce 2015 Complete Tests 1 e 2 AnswersDocument4 pagesFce 2015 Complete Tests 1 e 2 AnswersMárcia Bonfim83% (23)
- 21-Day Mind, Body and Spirit DetoxDocument6 pages21-Day Mind, Body and Spirit DetoxAjay TiwariNo ratings yet
- Cystic Fibrosis: Zoe Drullard and Avril GuillermoDocument18 pagesCystic Fibrosis: Zoe Drullard and Avril Guillermozoe drullardNo ratings yet
- Cystic Fibrosis: Gene TherapyDocument19 pagesCystic Fibrosis: Gene TherapyChansiekeiNo ratings yet
- Cystic FibrosisDocument3 pagesCystic Fibrosissumana yeasminNo ratings yet
- Cystic FibrosisDocument35 pagesCystic Fibrosisvani reddyNo ratings yet
- Cystic FibrosisDocument6 pagesCystic Fibrosischurchil owinoNo ratings yet
- The Cell Biology of Cystic FibrosisDocument23 pagesThe Cell Biology of Cystic FibrosisShamarie Love MaribaoNo ratings yet
- Theme1AppliedLecture2 BIO1A03Fall2022Document27 pagesTheme1AppliedLecture2 BIO1A03Fall2022proplayer910No ratings yet
- Cystic FibrosisDocument3 pagesCystic FibrosisstanscimagNo ratings yet
- Biochemistry Presentation.Document3 pagesBiochemistry Presentation.MIMIbako67No ratings yet
- Cystic FibrosisDocument14 pagesCystic FibrosisAishistic thingsNo ratings yet
- 16.5 Gene Therapy: 10.1 CoordinationDocument18 pages16.5 Gene Therapy: 10.1 Coordinationsohailnoreen5062No ratings yet
- Cystic Fibrosis: Bio FactsheetDocument3 pagesCystic Fibrosis: Bio FactsheetJavier SaadNo ratings yet
- Cystic Fibrosis: Dr. Pragasam Viswanathan Professor, SBSTDocument16 pagesCystic Fibrosis: Dr. Pragasam Viswanathan Professor, SBSTMaru Mengesha Worku 18BBT0285No ratings yet
- 1.3.7 Cystic Fibrosis: Chapter 3 Cystic Fibrosis HBIO1 - The Body and Its Diseases AQA AS Human BiologyDocument22 pages1.3.7 Cystic Fibrosis: Chapter 3 Cystic Fibrosis HBIO1 - The Body and Its Diseases AQA AS Human BiologyMaha NaserNo ratings yet
- The Genetics of Cystic FibrosisDocument24 pagesThe Genetics of Cystic FibrosisThrilok Nath MadduruNo ratings yet
- Cysticfibrosisdonebymyfriend 120218095057 Phpapp02Document28 pagesCysticfibrosisdonebymyfriend 120218095057 Phpapp02Elham AlwagaaNo ratings yet
- Cystic FibrosisDocument21 pagesCystic FibrosisGouthamNo ratings yet
- J-2.16 Bio1201Document7 pagesJ-2.16 Bio1201gert16No ratings yet
- CR Project On Cystic FibrosisDocument3 pagesCR Project On Cystic FibrosisMorgan PeggNo ratings yet
- Cystic FibrosisDocument9 pagesCystic FibrosiszarasarNo ratings yet
- Genes and Patterns of InheritanceDocument25 pagesGenes and Patterns of InheritanceNeelam HanifNo ratings yet
- Anaphy-Case Study-CjbascoDocument3 pagesAnaphy-Case Study-CjbascoChristine Jane BascoNo ratings yet
- Cystic Fibrosis Fact SheetDocument6 pagesCystic Fibrosis Fact SheetAnggun SafariantiniNo ratings yet
- Microsoft PowerPoint Presentation جديدDocument10 pagesMicrosoft PowerPoint Presentation جديدakram alrdayNo ratings yet
- Cystic Fibrosis With Case StudyDocument27 pagesCystic Fibrosis With Case StudyAviva Tuteja100% (1)
- ITW - Cystic Fibrosis Lecture 1Document9 pagesITW - Cystic Fibrosis Lecture 1Maha KhanNo ratings yet
- Cystic Fibrosis (CF) Final 1Document34 pagesCystic Fibrosis (CF) Final 1Mapis BravoNo ratings yet
- Cystic FibrosisDocument29 pagesCystic FibrosisAviva TutejaNo ratings yet
- Cystic FibrosisDocument22 pagesCystic FibrosisIeva PetrylaiteNo ratings yet
- Cystic FibrosisDocument19 pagesCystic FibrosisChristian Dene Opada TatoyNo ratings yet
- Genetic Disease ChartDocument1 pageGenetic Disease ChartNellena Kaye PedrigalNo ratings yet
- Case Study: Cystic Fibrosis: Manalo, Joanne Erica S. BS Respiratory TherapyDocument20 pagesCase Study: Cystic Fibrosis: Manalo, Joanne Erica S. BS Respiratory TherapyJohn Carlo ConsultaNo ratings yet
- A New Hope. Trikafta For The Treatment of Cystic FibrosisDocument6 pagesA New Hope. Trikafta For The Treatment of Cystic FibrosisTania CarreñoNo ratings yet
- CFTR GeneticsreviewDocument12 pagesCFTR GeneticsreviewTanya GreyNo ratings yet
- Diseases of Pattern of InheritanceDocument7 pagesDiseases of Pattern of InheritanceLABORATORY ASTMMCNo ratings yet
- Cystic FibrosisDocument23 pagesCystic FibrosisawirbshtiwanNo ratings yet
- Cystic FibrosisDocument71 pagesCystic Fibrosishumera0% (1)
- Anatomy of Cystic FibrosisDocument5 pagesAnatomy of Cystic FibrosislazylawatudentNo ratings yet
- Cystic Fibrosis: Nature Reviews Disease Primers May 2015Document20 pagesCystic Fibrosis: Nature Reviews Disease Primers May 2015Tapan AnkleshwariaNo ratings yet
- Cystic Fibrosis Pancreas: Deevon M. Cariaga FEU-NRMF MedicineDocument7 pagesCystic Fibrosis Pancreas: Deevon M. Cariaga FEU-NRMF MedicinedeevoncNo ratings yet
- Cystic FibrosisDocument13 pagesCystic FibrosisAnkita guptaNo ratings yet
- Ewence2020Cystic FibrosisDocument5 pagesEwence2020Cystic FibrosismacedovendezuNo ratings yet
- A Review of Trikafta: Triple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulator TherapyDocument7 pagesA Review of Trikafta: Triple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulator TherapySugi AntoNo ratings yet
- Cystic FibrosisDocument16 pagesCystic FibrosisAzzahra AzmiNo ratings yet
- Cystic FibrosisDocument33 pagesCystic FibrosisGreeshma ShajiNo ratings yet
- Cystic Fibrosis Research NotesDocument8 pagesCystic Fibrosis Research NotesNicole KrawitzNo ratings yet
- Cystic Fibrosis Case ReportDocument17 pagesCystic Fibrosis Case ReportMarjo100% (1)
- Columnaris: Manimozhi. E AAH-MA8-05 Aahm, CifeDocument30 pagesColumnaris: Manimozhi. E AAH-MA8-05 Aahm, CifeManimozhi EthirajanNo ratings yet
- Fibrosis Quística - NEJM02112023Document15 pagesFibrosis Quística - NEJM02112023Jesus A. Pineda GarciaNo ratings yet
- Cystic FibrosisDocument20 pagesCystic FibrosisIndrajit BaruaNo ratings yet
- Pemicu 5 Cessy ChristyDocument205 pagesPemicu 5 Cessy Christygrace liwantoNo ratings yet
- Cystic FibrosisDocument20 pagesCystic FibrosisIndrajit Barua100% (1)
- Cystic FibrosisDocument2 pagesCystic Fibrosisthatkid94No ratings yet
- Review of Rapid Advances in Cystic FibrosisDocument7 pagesReview of Rapid Advances in Cystic Fibrosismihaelaatanasova123No ratings yet
- Cystic FibrosisDocument32 pagesCystic FibrosisSrm Genetics100% (1)
- Cystic FibrosisDocument40 pagesCystic FibrosisMoonmoon SinhaNo ratings yet
- Adaptation To High Rates of Chromosomal Instability and Aneuploidy Through Multiple Pathways in Budding YeastDocument19 pagesAdaptation To High Rates of Chromosomal Instability and Aneuploidy Through Multiple Pathways in Budding YeastFernandoNo ratings yet
- Cysticfibrosis PDFDocument20 pagesCysticfibrosis PDFTORRES Hazel Ann I.No ratings yet
- CysticfibrosisDocument6 pagesCysticfibrosisSghir SghirNo ratings yet
- Organisational BehaviourDocument279 pagesOrganisational BehaviourSumitha SelvarajNo ratings yet
- The Literary Comparison Contrast EssayDocument7 pagesThe Literary Comparison Contrast EssayThapelo SebolaiNo ratings yet
- Conceptual Design of An Automotive Composite Brake Pedal: Mohd Sapuan Salit, Mohd Syed Ali Molla and MD Liakot AliDocument5 pagesConceptual Design of An Automotive Composite Brake Pedal: Mohd Sapuan Salit, Mohd Syed Ali Molla and MD Liakot Aliseran ünalNo ratings yet
- 2011 Diving Catalogue BEUCHATDocument60 pages2011 Diving Catalogue BEUCHATRalph KramdenNo ratings yet
- Third Party Logistics: A Literature Review and Research AgendaDocument26 pagesThird Party Logistics: A Literature Review and Research AgendaRuxandra Radus100% (1)
- Justin Bieber Is - Famous Singer.: (You Must Read Out The Whole Sentence.)Document2 pagesJustin Bieber Is - Famous Singer.: (You Must Read Out The Whole Sentence.)Irene De la FuenteNo ratings yet
- Cosmetics Case ProjectDocument16 pagesCosmetics Case ProjectLaura NardiNo ratings yet
- Reactor and Regenerator System of FCCDocument67 pagesReactor and Regenerator System of FCCDai NamNo ratings yet
- The DJ Test: Personalised Report and Recommendations For Alex YachevskiDocument34 pagesThe DJ Test: Personalised Report and Recommendations For Alex YachevskiSashadanceNo ratings yet
- CASHe Soa 568304-1Document2 pagesCASHe Soa 568304-1sathyaNo ratings yet
- Lab 6 Shouq PDFDocument10 pagesLab 6 Shouq PDFreemy100% (1)
- BSI BCI Horizon Scan Report 2016 PDFDocument32 pagesBSI BCI Horizon Scan Report 2016 PDFSandraNo ratings yet
- Small Unit Operations: PMA Core Values: Selfless Service To God and Country, Honor, ExcellenceDocument33 pagesSmall Unit Operations: PMA Core Values: Selfless Service To God and Country, Honor, ExcellenceAbay GeeNo ratings yet
- Full Download Art of Leadership 5th Edition Manning Test Bank PDF Full ChapterDocument36 pagesFull Download Art of Leadership 5th Edition Manning Test Bank PDF Full Chaptermasqueedenized8a43l100% (19)
- What Do You Know About Jobs - 28056Document2 pagesWhat Do You Know About Jobs - 28056Kadek DharmawanNo ratings yet
- Accounting Standards - Handwritten Notes - Udesh Regular - Group 1Document24 pagesAccounting Standards - Handwritten Notes - Udesh Regular - Group 1Uday TomarNo ratings yet
- Past Progressive SeptimoDocument1 pagePast Progressive Septimoferney cordobaNo ratings yet
- Bsl6 Qual Spec 11-12 FinalDocument35 pagesBsl6 Qual Spec 11-12 FinalBSLcourses.co.ukNo ratings yet
- Postmodern Nihilism Theory and LiteratureDocument336 pagesPostmodern Nihilism Theory and LiteratureCátia Tavares100% (3)
- Comparative and Superlative Adjectives PresentationDocument15 pagesComparative and Superlative Adjectives Presentationapi-248688582No ratings yet
- Code of Ethics For Professional TeachersDocument12 pagesCode of Ethics For Professional TeachersJean GuyuranNo ratings yet
- GST/HST Credit Application For Individuals Who Become Residents of CanadaDocument4 pagesGST/HST Credit Application For Individuals Who Become Residents of CanadaAndrea Dr FanisNo ratings yet
- PCA Churches in CaliforniaDocument4 pagesPCA Churches in CaliforniaLIGHT GNo ratings yet
- Deftones - Back To School (Pink Maggit Rap Version) : (Intro)Document2 pagesDeftones - Back To School (Pink Maggit Rap Version) : (Intro)zwartwerkerijNo ratings yet
- Effect of Bolt Pretension in Single Lap Bolted Joint IJERTV4IS010269Document4 pagesEffect of Bolt Pretension in Single Lap Bolted Joint IJERTV4IS010269ayush100% (1)
- Womens EssentialsDocument50 pagesWomens EssentialsNoor-uz-Zamaan AcademyNo ratings yet
- Petitioner vs. VS.: Third DivisionDocument12 pagesPetitioner vs. VS.: Third DivisionAnonymous QR87KCVteNo ratings yet