Download as pdf or txt
You might also like
- Neurology Multiple Choice Questions With Explanations: Volume IFrom EverandNeurology Multiple Choice Questions With Explanations: Volume IRating: 4 out of 5 stars4/5 (7)
- Amyotrophic Lateral SclerosisDocument58 pagesAmyotrophic Lateral SclerosisAathi PathmanathanNo ratings yet
- Fast Facts: Diagnosing Amyotrophic Lateral Sclerosis: Clinical wisdom to facilitate faster diagnosisFrom EverandFast Facts: Diagnosing Amyotrophic Lateral Sclerosis: Clinical wisdom to facilitate faster diagnosisNo ratings yet
- Clinical Anatomy of Upper Limb (KLM)Document4 pagesClinical Anatomy of Upper Limb (KLM)Ceo MedComNo ratings yet
- Clinical Features of Amyotrophic Lateral Sclerosis and Other Forms of Motor Neuron Disease UpToDate PDFDocument21 pagesClinical Features of Amyotrophic Lateral Sclerosis and Other Forms of Motor Neuron Disease UpToDate PDFThái BảoNo ratings yet
- Diagnosis of Amyotrophic Lateral Sclerosis and Other Forms of Motor Neuron Disease - UpToDateDocument34 pagesDiagnosis of Amyotrophic Lateral Sclerosis and Other Forms of Motor Neuron Disease - UpToDatehazalsariyildizNo ratings yet
- Esclerosis ArticuloDocument15 pagesEsclerosis ArticuloLauren BarretoNo ratings yet
- Pinto 2019Document12 pagesPinto 2019Fábio VinholyNo ratings yet
- Clinical Spectrum of Motor Neuron Disorders.8Document21 pagesClinical Spectrum of Motor Neuron Disorders.8Anonymous vnv6QFNo ratings yet
- Figure 1: Proposed Mechanisms Underlying Neurodegeneration in ALSDocument4 pagesFigure 1: Proposed Mechanisms Underlying Neurodegeneration in ALSJeroen MulderNo ratings yet
- Current Concepts About Motr Neuron DiseaseDocument68 pagesCurrent Concepts About Motr Neuron DiseaseMuhammad MuaazNo ratings yet
- Motor Neurone DiseaseDocument25 pagesMotor Neurone DiseasemoshiegonNo ratings yet
- Up To Date SLADocument13 pagesUp To Date SLAcrr r r jNo ratings yet
- FGDDFGDGDGDocument26 pagesFGDDFGDGDGiroxikajNo ratings yet
- Motor Neuron DiseaseDocument7 pagesMotor Neuron DiseasegeraldineongNo ratings yet
- Amyotrophic Lateral SclerosisDocument11 pagesAmyotrophic Lateral SclerosisAnomalie12345No ratings yet
- Amyotrophic Lateral SclerosisDocument23 pagesAmyotrophic Lateral SclerosisAfshan Pt T100% (1)
- Amyotrophic Lateral SclerosisDocument15 pagesAmyotrophic Lateral SclerosisYakan AbdulrahmanNo ratings yet
- American Academy of Neurology Key PointsDocument193 pagesAmerican Academy of Neurology Key Pointszayat13No ratings yet
- Clinical and Genetic Basis of Familial Amyotrophic Lateral Sclerosis (Revisión)Document12 pagesClinical and Genetic Basis of Familial Amyotrophic Lateral Sclerosis (Revisión)Francisco Ahumada MéndezNo ratings yet
- Motor Neuron Disease - StatPearls - NCBI BookshelfDocument16 pagesMotor Neuron Disease - StatPearls - NCBI BookshelfAmr BashaNo ratings yet
- Spasticity and ManagementDocument15 pagesSpasticity and ManagementMasa MasaNo ratings yet
- Neuron Diseases, Which Are Characterized by The GradualDocument12 pagesNeuron Diseases, Which Are Characterized by The GradualTanu ShreeNo ratings yet
- Amyotrophic Lateral SclerosisDocument2 pagesAmyotrophic Lateral SclerosisErwin Maulana Farmanda PutraNo ratings yet
- ALS Brochure 508compDocument24 pagesALS Brochure 508compidaayupranitaNo ratings yet
- Amyotrophic Lateral SclerosisDocument9 pagesAmyotrophic Lateral SclerosisCaroline ItnerNo ratings yet
- Atrfoia Espinal MioclonicDocument18 pagesAtrfoia Espinal MioclonicmmblozsnoNo ratings yet
- Amyotrophic Lateral Sclerosis, Thesis M1, Version 2, 2024Document24 pagesAmyotrophic Lateral Sclerosis, Thesis M1, Version 2, 2024Ghina GhiehNo ratings yet
- Overview of The Hereditary Ataxias - UpToDateDocument15 pagesOverview of The Hereditary Ataxias - UpToDatericanoy191No ratings yet
- Focal Epilepsy Causes and Clinical Features - UpToDateDocument44 pagesFocal Epilepsy Causes and Clinical Features - UpToDateBenjamínGalvanNo ratings yet
- Amyotrophic Lateral Sclerosis IDocument18 pagesAmyotrophic Lateral Sclerosis IAlegna AhsramNo ratings yet
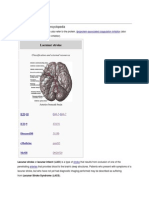
- Lacunar StrokeDocument5 pagesLacunar StrokeLindsay NolanNo ratings yet
- ALS - A Clinical ReviewDocument26 pagesALS - A Clinical ReviewAurelia Sartika TongkuNo ratings yet
- Brooks 2000Document8 pagesBrooks 2000Daisy Jennifer Paulino AriasNo ratings yet
- Clinical EMG ALSDocument7 pagesClinical EMG ALSAbul Shamsud DoulahNo ratings yet
- Muscular DystoniaDocument20 pagesMuscular Dystoniarudresh singhNo ratings yet
- What Is Amyotrophic Lateral Sclerosis?Document4 pagesWhat Is Amyotrophic Lateral Sclerosis?Amoza MonsaleNo ratings yet
- Amyotrophic Lateral SclerosisDocument8 pagesAmyotrophic Lateral SclerosisridwanNo ratings yet
- Diseases of Spine and Spinal Cord: D R. Devi Wuysang, M.Si.,Sp.SDocument41 pagesDiseases of Spine and Spinal Cord: D R. Devi Wuysang, M.Si.,Sp.Sanugerah100% (1)
- Myoclonus With DementiaDocument8 pagesMyoclonus With DementiaSantosh DashNo ratings yet
- ABSTRACT-DIAGNOSTIC APPROACH OF AMYOTROPHIC LATERAL SCLEROSIS IN DEVELOPING COUNTRY-A CASE REPORT Perbaikan - InggrisDocument1 pageABSTRACT-DIAGNOSTIC APPROACH OF AMYOTROPHIC LATERAL SCLEROSIS IN DEVELOPING COUNTRY-A CASE REPORT Perbaikan - InggrisMufliha IlhamNo ratings yet
- Degenerative Diseases of The Nervous SystemDocument87 pagesDegenerative Diseases of The Nervous SystemNancy MosesNo ratings yet
- Motor DisordersDocument7 pagesMotor DisordersDian AzhariaNo ratings yet
- Spinocerebellar AtaxiaDocument3 pagesSpinocerebellar AtaxiaSeth LyhalimNo ratings yet
- Research Paper On Amyotrophic Lateral SclerosisDocument5 pagesResearch Paper On Amyotrophic Lateral Sclerosismzgxwevkg100% (1)
- The Pathophysiology of Alzheimer'S Disease and Directions in TreatmentDocument15 pagesThe Pathophysiology of Alzheimer'S Disease and Directions in TreatmentEnerolisa ParedesNo ratings yet
- Sdarticle PDFDocument9 pagesSdarticle PDFManjeev GuragainNo ratings yet
- Multifocal Motor NeuropathyDocument11 pagesMultifocal Motor Neuropathymedana5No ratings yet
- Chapter 1Document25 pagesChapter 1CharleneKronstedtNo ratings yet
- Em - Revision Del Lancet-1Document16 pagesEm - Revision Del Lancet-1César Vásquez AguilarNo ratings yet
- 401 2017 Article 1708Document23 pages401 2017 Article 1708HALLYSSON RIBEIRO DA SILVANo ratings yet
- Motor Symptoms and Schizophrenia: NeuropsychobiologyDocument17 pagesMotor Symptoms and Schizophrenia: Neuropsychobiologymuhammad fahrizaNo ratings yet
- Practical Histology:) Motor Neurone Disease (MNDDocument5 pagesPractical Histology:) Motor Neurone Disease (MNDعلي حسين عودة العلياويNo ratings yet
- Neuropsychological Assessment in Different King's Clinical Stages of Amyotrophic Lateral SclerosisDocument9 pagesNeuropsychological Assessment in Different King's Clinical Stages of Amyotrophic Lateral SclerosisLlamadme ImanolNo ratings yet
- Leg Weakness in A 66-Year-Old Woman: A Common Presentation of An Uncommon DiseaseDocument10 pagesLeg Weakness in A 66-Year-Old Woman: A Common Presentation of An Uncommon DiseaseCarlos De Santiago CastilloNo ratings yet
- Motor Neurone Disease ThesisDocument2 pagesMotor Neurone Disease ThesisParth Vijay SinghNo ratings yet
- Peripheral Nervous System Disorders Author EknygosDocument25 pagesPeripheral Nervous System Disorders Author EknygosPredrag NikolicNo ratings yet
- Upper Motor Neuron DiseasesDocument7 pagesUpper Motor Neuron DiseasesRyl WonNo ratings yet
- Amyotrophic Lateral Sclerosis A.K.A Lou Gehrig's Disease: Bobbette Miller DPT, NCSDocument14 pagesAmyotrophic Lateral Sclerosis A.K.A Lou Gehrig's Disease: Bobbette Miller DPT, NCSJohn SmithNo ratings yet
- Amyotrophic Lateral SclerosisDocument3 pagesAmyotrophic Lateral Sclerosisclumsy16No ratings yet
- 0004 282X Anp 72 06 451Document6 pages0004 282X Anp 72 06 451Okhuarobo SamuelNo ratings yet
- NCM 112 TH (12F)Document24 pagesNCM 112 TH (12F)Justine April YbanezNo ratings yet
- Written Assessment Information Booklet Website July 2018Document11 pagesWritten Assessment Information Booklet Website July 2018aaditi2mudaliarNo ratings yet
- 1 Q PDFDocument44 pages1 Q PDFMysheb SSNo ratings yet
- Krok 2 Dentistry MCQsDocument536 pagesKrok 2 Dentistry MCQsRachid HissaneNo ratings yet
- Interpretations: How To Use Faecal Elastase TestingDocument6 pagesInterpretations: How To Use Faecal Elastase TestingguschinNo ratings yet
- Syllabus Assistant Surgeon Exam PDFDocument9 pagesSyllabus Assistant Surgeon Exam PDFMaguNo ratings yet
- Las 1 Health 7 Q4Document8 pagesLas 1 Health 7 Q4arlenejoy.donadilloNo ratings yet
- Case PBL ICM2 Week 3 Jumat-TSDocument33 pagesCase PBL ICM2 Week 3 Jumat-TSsylvia haryantoNo ratings yet
- The Cellular Basis of Disease: Cell Injury 3 Apoptosis and Necrosis Cellular AgingDocument49 pagesThe Cellular Basis of Disease: Cell Injury 3 Apoptosis and Necrosis Cellular AgingZabella SilvianaNo ratings yet
- Tetanus (Clostridium Tetani)Document29 pagesTetanus (Clostridium Tetani)ped medNo ratings yet
- How I Treat Anemia in Pregnancy: Iron, Cobalamin, and FolateDocument11 pagesHow I Treat Anemia in Pregnancy: Iron, Cobalamin, and FolateAgungBudiPamungkasNo ratings yet
- Rickets, Osteomalacia and Renal OsteodystrophyDocument13 pagesRickets, Osteomalacia and Renal Osteodystrophyesther_aprilNo ratings yet
- Small Animal Emergency and Critical Care MedicineDocument193 pagesSmall Animal Emergency and Critical Care MedicineYaiza Garcia CasadoNo ratings yet
- Oncology Drills With Answers and RationalesDocument41 pagesOncology Drills With Answers and RationalesCarol Kayas100% (1)
- Solution Manual For Investigations in Environmental Geology, 3/E 3Rd Edition Duncan D. Foley, Garry D. Mckenzie, Russell O. UtgardDocument31 pagesSolution Manual For Investigations in Environmental Geology, 3/E 3Rd Edition Duncan D. Foley, Garry D. Mckenzie, Russell O. Utgardraymond.nelson503100% (7)
- Case Study - ARDS Due To COVID19Document4 pagesCase Study - ARDS Due To COVID19Melody B. MiguelNo ratings yet
- Little Mermaid 1Document12 pagesLittle Mermaid 1Marsh MallowNo ratings yet
- MenopauseDocument32 pagesMenopauseAdefa Shiferaw100% (1)
- Kussmaul BreathingDocument3 pagesKussmaul BreathingsakuraleeshaoranNo ratings yet
- Kode Icd 10Document536 pagesKode Icd 10AkbarisArisNo ratings yet
- BIOLOGY PROJECT (Class 12)Document21 pagesBIOLOGY PROJECT (Class 12)Manoghna MaheshNo ratings yet
- EmergencyDocument86 pagesEmergencyyazzNo ratings yet
- Anti Seizure DrugsDocument76 pagesAnti Seizure DrugsMwanja Moses100% (1)
- Time Specific Objectives Teaching, Learning Activities EvaluationDocument13 pagesTime Specific Objectives Teaching, Learning Activities EvaluationDeepti KukretiNo ratings yet
- Senile DementiaDocument11 pagesSenile DementiaSubha MaheswariNo ratings yet
- 2023 ESH Guidelines For The Management of Arterial HypertensionDocument199 pages2023 ESH Guidelines For The Management of Arterial HypertensionErika Jiménez De LaraNo ratings yet
- By Dr. Smitha B K Post Graduate Student Department of Preventive and Community DentistryDocument58 pagesBy Dr. Smitha B K Post Graduate Student Department of Preventive and Community DentistryAnnupriya KhannaNo ratings yet
- Chapter Practice Test: Home Chapter 16: The Endocrine SystemDocument4 pagesChapter Practice Test: Home Chapter 16: The Endocrine SystemHUAWEI HUAWEINo ratings yet
- Vaughan and Asburys General Ophthalmology 19E 2018Document11 pagesVaughan and Asburys General Ophthalmology 19E 2018FebeNo ratings yet