Download as pdf or txt
You might also like
- Dawn Bradley Berry - Domestic Violence Source Book-Lowell House (1998)Document566 pagesDawn Bradley Berry - Domestic Violence Source Book-Lowell House (1998)mohd amees100% (1)
- Sumit Sarkar - Tanika Sarkar - Women and Social Reform in Modern India - A Reader Vol 1. 1Document476 pagesSumit Sarkar - Tanika Sarkar - Women and Social Reform in Modern India - A Reader Vol 1. 1mohd amees100% (1)
- Dissolution Enhancement and Formulation of Film CoDocument12 pagesDissolution Enhancement and Formulation of Film CobimaNo ratings yet
- Chemical Engineering Journal: SciencedirectDocument11 pagesChemical Engineering Journal: SciencedirectOussama El BouadiNo ratings yet
- Formulation and Evaluation of Nanoemulsion For Solubility Enhancement of KetoconazoleDocument14 pagesFormulation and Evaluation of Nanoemulsion For Solubility Enhancement of KetoconazoledgdNo ratings yet
- Budhi 2018 IOP Conf. Ser. Earth Environ. Sci. 105 012063Document7 pagesBudhi 2018 IOP Conf. Ser. Earth Environ. Sci. 105 012063nafsiyah xyzNo ratings yet
- International Journal of Pharmaceutics: A B B B B B BDocument10 pagesInternational Journal of Pharmaceutics: A B B B B B BVizay R. AlieNo ratings yet
- 125941119Document4 pages125941119Trung HoàngNo ratings yet
- Chế tạo và mô tả đặc tính của các hạt nano Fucoidan-Chitosan bằng phương pháp siêu âmDocument6 pagesChế tạo và mô tả đặc tính của các hạt nano Fucoidan-Chitosan bằng phương pháp siêu âmTrang ThùyNo ratings yet
- Novel Spray Freeze-Drying Technique Using Four-Fluid NozzleDocument6 pagesNovel Spray Freeze-Drying Technique Using Four-Fluid NozzleApoorva KNo ratings yet
- Chitosan PapersDocument19 pagesChitosan PapersJayNo ratings yet
- UltrafiltrationDocument5 pagesUltrafiltrationYash PatelNo ratings yet
- 2013 - Inulina FarmaceuticaDocument8 pages2013 - Inulina FarmaceuticaKhrisThian LópezNo ratings yet
- ResearchDocument9 pagesResearchSalah Farhan NoriNo ratings yet
- JDRD164Document5 pagesJDRD164Julia MalikaNo ratings yet
- Preparation of Cassava Starch Nanoparticles and Their Application As A Carrier System For Curcumin DeliveryDocument9 pagesPreparation of Cassava Starch Nanoparticles and Their Application As A Carrier System For Curcumin DeliveryKool LokeshNo ratings yet
- Cellulose Nanofibers Produced From Banana Peel by Enzymatic Treatment Study of Process ConditionsDocument11 pagesCellulose Nanofibers Produced From Banana Peel by Enzymatic Treatment Study of Process ConditionsmusicontheroaddNo ratings yet
- Nanomedicine & NanotechnologyDocument5 pagesNanomedicine & NanotechnologyfikriNo ratings yet
- 2020 APT ElectrosprayDocument14 pages2020 APT ElectrosprayDian Ahmad HapidinNo ratings yet
- 630 1197 1 SMDocument8 pages630 1197 1 SMGotsvnNo ratings yet
- 10 1016@j Ejps 2019 104981Document9 pages10 1016@j Ejps 2019 104981sssNo ratings yet
- Hasil Penelitian PvaDocument7 pagesHasil Penelitian PvaEga TrikuntiantiNo ratings yet
- Stan 2019Document9 pagesStan 2019Septian Perwira YudhaNo ratings yet
- PharmageneDocument5 pagesPharmageneNuurus Sa'adahNo ratings yet
- Nano Particulas en Diseño de Formas FarmaceuticasDocument8 pagesNano Particulas en Diseño de Formas FarmaceuticasMonica Andrea Marulanda GuzmanNo ratings yet
- Journal of Molecular Liquids: Manas Kumar Guria, Medha Majumdar, Maitree BhattacharyyaDocument9 pagesJournal of Molecular Liquids: Manas Kumar Guria, Medha Majumdar, Maitree BhattacharyyaMaría Alejandra AyudeNo ratings yet
- Noble Metallic Nanoparticles From Waste Nypa Fruticans Fruit Husk - Biosynthesis Characterization Antibacterial Activity and Recyclable CatalysisDocument14 pagesNoble Metallic Nanoparticles From Waste Nypa Fruticans Fruit Husk - Biosynthesis Characterization Antibacterial Activity and Recyclable Catalysisratno TimurNo ratings yet
- Compaction Processor Pre - CompresionDocument9 pagesCompaction Processor Pre - CompresionADVOCATE ASHUTOSH SHARMANo ratings yet
- Novel Herbal Drug Microsphere Types of Preparation Characterization and Application A ReviewDocument5 pagesNovel Herbal Drug Microsphere Types of Preparation Characterization and Application A ReviewEditor IJTSRDNo ratings yet
- KT Hình Cầu Của ItraconazoleDocument11 pagesKT Hình Cầu Của ItraconazoleDương TrungNo ratings yet
- Nanosilica For Sustained Release of Quercetin and Its Antioxidant ActivityDocument10 pagesNanosilica For Sustained Release of Quercetin and Its Antioxidant ActivityAnonymous izrFWiQNo ratings yet
- Jelena Prvi 1-S2.0-S0927775717306192-Main PDFDocument7 pagesJelena Prvi 1-S2.0-S0927775717306192-Main PDFAlejandra Cale RadowitzNo ratings yet
- +01 +Ira+Desri+Rahmi+Copy+EditingDocument18 pages+01 +Ira+Desri+Rahmi+Copy+EditingDwi SetyaningsihNo ratings yet
- Vanistagar Dan SubhashiniDocument11 pagesVanistagar Dan SubhashinikajiNo ratings yet
- Kumar Vol1 Pag 33 41Document10 pagesKumar Vol1 Pag 33 41Sampath KumarNo ratings yet
- Chemical Composition and Properties of Maple Sap Treated With An Ultra High Membrane Concentration ProcessDocument9 pagesChemical Composition and Properties of Maple Sap Treated With An Ultra High Membrane Concentration ProcessDiego ViniciusNo ratings yet
- Setyaningsih 2018 IOP Conf. Ser. - Earth Environ. Sci. 141 012027Document9 pagesSetyaningsih 2018 IOP Conf. Ser. - Earth Environ. Sci. 141 012027Nabil FahmiNo ratings yet
- One-Pot Nanofibrillation of Cellulose and Nanocomposite Production in A Twin-Screw ExtruderDocument10 pagesOne-Pot Nanofibrillation of Cellulose and Nanocomposite Production in A Twin-Screw ExtruderHidayah AriffinNo ratings yet
- Aspirin Loaded Albumin Nanoparticles by Coacervation: Implications in Drug DeliveryDocument10 pagesAspirin Loaded Albumin Nanoparticles by Coacervation: Implications in Drug DeliveryafandianddonkeyNo ratings yet
- Spectrophotometric Determination and Commercial Formulation Oftebuconazole Fungicide After Derivatization 2150 3494 1000133Document4 pagesSpectrophotometric Determination and Commercial Formulation Oftebuconazole Fungicide After Derivatization 2150 3494 1000133lox agencyNo ratings yet
- Heliyon: Ilma Nugrahani, Winni Nur AuliDocument9 pagesHeliyon: Ilma Nugrahani, Winni Nur AuliNAVARRETE RODRIGUEZ ANGELICA MARIANo ratings yet
- Habit Kristal SolventscreeningandcrystalhabitofmetforminhydrochlorideDocument11 pagesHabit Kristal SolventscreeningandcrystalhabitofmetforminhydrochlorideLaksmi DwiNo ratings yet
- Studies On Formulation and In-Vitro Evaluation of Mouth Dissolving Tablets Containing Telmisartan by Using Box-Benkhen DesignDocument7 pagesStudies On Formulation and In-Vitro Evaluation of Mouth Dissolving Tablets Containing Telmisartan by Using Box-Benkhen DesignInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Filtration of Apple Juice by Nylon Nanofibrous MembranesDocument7 pagesFiltration of Apple Juice by Nylon Nanofibrous MembranesDavid CamargoNo ratings yet
- Characteristics Testing of Microcrystalline Cellulose From Nata de Coco Compared To Avicel PH 101 and Avicel PH 102Document5 pagesCharacteristics Testing of Microcrystalline Cellulose From Nata de Coco Compared To Avicel PH 101 and Avicel PH 102mela pertiwiNo ratings yet
- 2016 - Der Pharmacia Lettre (MSM)Document6 pages2016 - Der Pharmacia Lettre (MSM)Joel CNo ratings yet
- Formulation and Evaluation of Microemulsion Based Topical Hydrogel Containing LornoxicamDocument8 pagesFormulation and Evaluation of Microemulsion Based Topical Hydrogel Containing LornoxicamsafrinpujiNo ratings yet
- CocrystalDocument8 pagesCocrystaljokonudiNo ratings yet
- Identification Characterization and Evaluation ofDocument10 pagesIdentification Characterization and Evaluation ofAkhmad Rafi'iNo ratings yet
- Ae-Extracción de Adn Revelado Con Electroforesis de Métodos de Extracción de Adn y PCR de Dna de SuelosDocument7 pagesAe-Extracción de Adn Revelado Con Electroforesis de Métodos de Extracción de Adn y PCR de Dna de SuelosStefhany ValdeiglesiasNo ratings yet
- JYoungPharm 10 2 s84Document3 pagesJYoungPharm 10 2 s84panim130No ratings yet
- Natural Surfactants For Flotation Deinking in Paper RecyclingDocument12 pagesNatural Surfactants For Flotation Deinking in Paper RecyclingARUNPRASADEEENo ratings yet
- Colloid and Interface Science Communications: SciencedirectDocument8 pagesColloid and Interface Science Communications: SciencedirectOsneider Peña CuetoNo ratings yet
- The Application of Biological Processes in Several Industrial Wastewater in IndonesiaDocument48 pagesThe Application of Biological Processes in Several Industrial Wastewater in IndonesiaHasriyani HafidNo ratings yet
- Finasteride Loaded Nano Lipidic Carriers For Follicular Drug Deliver - 2022 - HeDocument10 pagesFinasteride Loaded Nano Lipidic Carriers For Follicular Drug Deliver - 2022 - Hepo hongNo ratings yet
- Biosynthesis of Eco-Friendly Silver Nano-Particles: The Efficiency of Fresh Leaves and Dried Leaves in The Synthesis of Silver NanoparticlesDocument13 pagesBiosynthesis of Eco-Friendly Silver Nano-Particles: The Efficiency of Fresh Leaves and Dried Leaves in The Synthesis of Silver NanoparticlesEditor IJTSRDNo ratings yet
- Journal of Molecular Structure: T. Ngulube, J.R. Gumbo, V. Masindi, A. MaityDocument11 pagesJournal of Molecular Structure: T. Ngulube, J.R. Gumbo, V. Masindi, A. Maityall green associatesNo ratings yet
- 1 s2.0 S0927776521000485 MainDocument10 pages1 s2.0 S0927776521000485 MainPreyansh SrivastavaNo ratings yet
- IJRPR20553Document5 pagesIJRPR20553AndhiNo ratings yet
- Hydrate Formation Wet Granulation Taylor LSDocument11 pagesHydrate Formation Wet Granulation Taylor LSDaniel Mateo OrtizNo ratings yet
- Shiv IJCRRDocument10 pagesShiv IJCRRnavodaya9No ratings yet
- Chemical Modification of Solid Surfaces by the Use of AdditivesFrom EverandChemical Modification of Solid Surfaces by the Use of AdditivesNo ratings yet
- Scarred For Life: A Review of Cesarean Section Scar Pregnancy and Potential Pitfalls in DiagnosisDocument12 pagesScarred For Life: A Review of Cesarean Section Scar Pregnancy and Potential Pitfalls in Diagnosismohd ameesNo ratings yet
- Bbac 386Document14 pagesBbac 386mohd ameesNo ratings yet
- A Tale of Two SummitDocument3 pagesA Tale of Two Summitmohd ameesNo ratings yet
- Wong 2017Document26 pagesWong 2017mohd ameesNo ratings yet
- 11 Just Q383Document29 pages11 Just Q383mohd ameesNo ratings yet
- JMI Multimedia CollectionDocument84 pagesJMI Multimedia Collectionmohd ameesNo ratings yet
- NYT+OP Jindal Global University Launch GuidebookDocument41 pagesNYT+OP Jindal Global University Launch Guidebookmohd ameesNo ratings yet
- Cup Hss Journals Collection 2021: Sno Code TitleDocument42 pagesCup Hss Journals Collection 2021: Sno Code Titlemohd ameesNo ratings yet
- 2FB-510 BZDocument1 page2FB-510 BZIvanslzrNo ratings yet
- Experiment 4Document20 pagesExperiment 4William Allan Arcilla100% (3)
- Plumbing Arithmetic ReviewerDocument69 pagesPlumbing Arithmetic Revieweryaoi yuriNo ratings yet
- Panasonic Manual SM - Cs-E18.21ckeDocument100 pagesPanasonic Manual SM - Cs-E18.21ckeaaNo ratings yet
- Digital Assignment-2 Process Automation and Control Lab LAB SLOT:-L33+L34Document32 pagesDigital Assignment-2 Process Automation and Control Lab LAB SLOT:-L33+L34Goutham KrishnaNo ratings yet
- C04 Calculus TopicsDocument90 pagesC04 Calculus TopicsAzizmanvaNo ratings yet
- EE201 Circuits N NetworksDocument2 pagesEE201 Circuits N NetworksArya ANo ratings yet
- 15 1D Barrier ProblemsDocument23 pages15 1D Barrier ProblemsengshimaaNo ratings yet
- Wait It Out - Larry NivenDocument6 pagesWait It Out - Larry NivenDaniel Colucci100% (1)
- LS1 - Analysis of Air-Conditioning Processes PDFDocument4 pagesLS1 - Analysis of Air-Conditioning Processes PDFdildin01No ratings yet
- SSMET2014 Pollara Improved Thermal Vacuum Chamber Temperature Performance Via Gaseous Nitrogen Thermal Conditioning UnitsDocument4 pagesSSMET2014 Pollara Improved Thermal Vacuum Chamber Temperature Performance Via Gaseous Nitrogen Thermal Conditioning Unitsbahloul mohamedNo ratings yet
- Questions PV Design 2023Document8 pagesQuestions PV Design 2023Mutaz M BanatNo ratings yet
- 2011 SkyScan Bruker 1176 X-Ray Microtomograph ManualDocument103 pages2011 SkyScan Bruker 1176 X-Ray Microtomograph ManualrhinemineNo ratings yet
- Short Length Timber & Advanced Timber Construction: Reference BooksDocument48 pagesShort Length Timber & Advanced Timber Construction: Reference Bookssanket pawarNo ratings yet
- Ejms 669Document9 pagesEjms 669unlaber zzNo ratings yet
- Soil Investigation Research PaperDocument5 pagesSoil Investigation Research Paperefgncpe8100% (3)
- P Delta PDFDocument14 pagesP Delta PDFRekanNo ratings yet
- Classwork or Homework Investigating Diffusion in AirDocument2 pagesClasswork or Homework Investigating Diffusion in AirPranav BISUMBHERNo ratings yet
- Simple Harmonic MotionDocument61 pagesSimple Harmonic Motionyn KimNo ratings yet
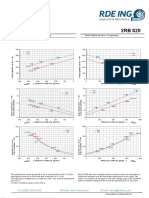
- Ficha Tecnica 2RB-820-7HH36Document2 pagesFicha Tecnica 2RB-820-7HH36Ronald Chacón QuirósNo ratings yet
- Pneumatic SchematicsDocument15 pagesPneumatic SchematicsAhmad HamoudaNo ratings yet
- Torque On A Stick LabDocument2 pagesTorque On A Stick LabAbdulMalik AriyoNo ratings yet
- Modeling of Solar/wind Hybrid Energy System Using MTALAB SimulinkDocument4 pagesModeling of Solar/wind Hybrid Energy System Using MTALAB SimulinkMahar Nadir AliNo ratings yet
- Pradhan Mantri Awas Yojana Housing For All (Urban) in AP StateDocument120 pagesPradhan Mantri Awas Yojana Housing For All (Urban) in AP StateKishore Nayak kNo ratings yet
- 10.6 AnswersDocument19 pages10.6 AnswersDiksha PatelNo ratings yet
- M0707 Acabados SuperficialDocument29 pagesM0707 Acabados SuperficialUbaldo Garcia ZaragozaNo ratings yet
- Planning A Code-Compliant, Off-Grid PV System: John WilesDocument6 pagesPlanning A Code-Compliant, Off-Grid PV System: John WilesDaniel MilosevskiNo ratings yet
- Objectives: The Nature of MatterDocument31 pagesObjectives: The Nature of MatterJoanna Marie R. PeranteNo ratings yet
- Design Guide 9 Revisions and Errata ListDocument5 pagesDesign Guide 9 Revisions and Errata ListFateh QuadriNo ratings yet
- Q173D (S) CPU/Q172D (S) CPU Motion Controller Programming Manual (Safety Observation)Document252 pagesQ173D (S) CPU/Q172D (S) CPU Motion Controller Programming Manual (Safety Observation)Anh Tuân LeNo ratings yet