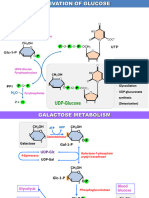
Download as pdf or txt
You might also like
- Galactosemia by Group 9Document41 pagesGalactosemia by Group 9Sree Balaji Srinivas Katakam100% (1)
- GalactosemiaDocument3 pagesGalactosemiaChiatsuNo ratings yet
- Changing Focus of Care in Community Mental HealthDocument13 pagesChanging Focus of Care in Community Mental HealthSyed Hussain89% (9)
- GALT Defi Ciency: GalactosemiaDocument8 pagesGALT Defi Ciency: GalactosemiaI'Jaz Farritz MuhammadNo ratings yet
- Gangguan Metabolisme KarbohidratDocument17 pagesGangguan Metabolisme KarbohidratDyah Ayu Kusumo WinahyuNo ratings yet
- Galactosemia - Scriver. MMBID 2006Document92 pagesGalactosemia - Scriver. MMBID 2006BarDFNo ratings yet
- Test Information GALEDocument5 pagesTest Information GALEcaycee rosarioNo ratings yet
- GalactosemiaDocument41 pagesGalactosemiaArlene DaroNo ratings yet
- GALACTOSEMIAlabDocument49 pagesGALACTOSEMIAlabShen VillamilNo ratings yet
- Biochemistry, Molecular Biology and Molecular Genetics of Galactosemia (PDFDrive)Document23 pagesBiochemistry, Molecular Biology and Molecular Genetics of Galactosemia (PDFDrive)Eng CirroNo ratings yet
- Galactose MiaDocument12 pagesGalactose MiaKaren EstavilloNo ratings yet
- GalectoseamiaDocument10 pagesGalectoseamiaomersalah1982No ratings yet
- Galactosemia by SlidesgoDocument24 pagesGalactosemia by SlidesgoCherie ThompsonNo ratings yet
- Provided by Liberty University Digital CommonsDocument32 pagesProvided by Liberty University Digital CommonsWessam WailNo ratings yet
- Galactosemia An InsightDocument5 pagesGalactosemia An InsightVineet MariyappanNo ratings yet
- 5 Galactose Fructose - PPT 2016Document6 pages5 Galactose Fructose - PPT 2016lou765500No ratings yet
- Monosaccharides MetabolismDocument28 pagesMonosaccharides MetabolismramaNo ratings yet
- Articulo 1 GalactosemiaDocument28 pagesArticulo 1 GalactosemiaIsabella CarbonariNo ratings yet
- Galactosemia: Prepared by Group VDocument36 pagesGalactosemia: Prepared by Group VVeyari RamirezNo ratings yet
- Nutrients 15 00010Document12 pagesNutrients 15 00010matejNo ratings yet
- Disorders of Carbohydrates Metabolism MetabolismDocument21 pagesDisorders of Carbohydrates Metabolism MetabolismUsman Ali AkbarNo ratings yet
- Biomolecules 12 00968Document14 pagesBiomolecules 12 00968matejNo ratings yet
- Galactose Tolerance TestDocument11 pagesGalactose Tolerance TestVirat Singh83% (6)
- Current and Future Treatment of Galactosemia - Journal of Personalized MedicineDocument15 pagesCurrent and Future Treatment of Galactosemia - Journal of Personalized MedicineChalida HayulaniNo ratings yet
- GalactosemiaDocument6 pagesGalactosemiaakbar alituNo ratings yet
- Insulin Secretion - Newer PerspectiveDocument6 pagesInsulin Secretion - Newer PerspectivehhhNo ratings yet
- GalactitolDocument8 pagesGalactitolHumayun Rashid ShakilNo ratings yet
- Differential DiagnosesDocument2 pagesDifferential DiagnosesJeremy EvansNo ratings yet
- Glycolysis: DR Imran SiddiquiDocument13 pagesGlycolysis: DR Imran Siddiquiapi-19824406No ratings yet
- LO GalactosemiaDocument2 pagesLO GalactosemiaJeremy EvansNo ratings yet
- Biochemistry ' Section Ii: Electron Transport Chain and Oxidative PhosphorylationDocument6 pagesBiochemistry ' Section Ii: Electron Transport Chain and Oxidative PhosphorylationAvanca LouisNo ratings yet
- Galactose Metabolism in Saccharomyces CerevisiaeDocument12 pagesGalactose Metabolism in Saccharomyces CerevisiaeFarida RahayuNo ratings yet
- Inserto Gluc3 HK RocheDocument4 pagesInserto Gluc3 HK RocheBruno ValeraNo ratings yet
- 1 GluconeogenesisDocument31 pages1 Gluconeogenesisemery100% (1)
- Azerbaijan Medical UniversityDocument52 pagesAzerbaijan Medical UniversityDr ZakaryaNo ratings yet
- En GlucoseDocument4 pagesEn GlucosemsaidsaidyoussefNo ratings yet
- Enzyme-Based Amperometric Galactose Biosensors: A ReviewDocument9 pagesEnzyme-Based Amperometric Galactose Biosensors: A ReviewCezaraDianaNo ratings yet
- GalactosemiaDocument18 pagesGalactosemiaHenil SavaliyaNo ratings yet
- Biochem SuperTableDocument2 pagesBiochem SuperTablePrincess MarielleNo ratings yet
- Classic GalactosemiaDocument18 pagesClassic GalactosemiaHelena QuintNo ratings yet
- Penumonia and COPDDocument50 pagesPenumonia and COPDDr ZakaryaNo ratings yet
- Galactosemia and Lactose Intolerance Powerpoint FinalDocument72 pagesGalactosemia and Lactose Intolerance Powerpoint FinalNico Batalla60% (5)
- 3-HMP and Glucuronic Acid PathwaysDocument40 pages3-HMP and Glucuronic Acid Pathwayslou765500No ratings yet
- K-Lolac DataDocument19 pagesK-Lolac DataShruthiNo ratings yet
- Biochem SuperTable PDFDocument2 pagesBiochem SuperTable PDFPrincess MarielleNo ratings yet
- The Gallic Acid-Phospholipid Complex Improved The Antioxidant Potential of Gallic Acid by Enhancing Its BioavailabilityDocument9 pagesThe Gallic Acid-Phospholipid Complex Improved The Antioxidant Potential of Gallic Acid by Enhancing Its BioavailabilityHoàng Sơn Nguyễn LêNo ratings yet
- A Novel Therapeutic Agent For Type 2 Diabetes Mellitus: SGLT2 InhibitorDocument13 pagesA Novel Therapeutic Agent For Type 2 Diabetes Mellitus: SGLT2 InhibitorSheldon SilvaNo ratings yet
- Wei LinDocument9 pagesWei Linasma.almhsnNo ratings yet
- GALACTOSEMIADocument13 pagesGALACTOSEMIANorfazilah ZainulNo ratings yet
- Glycogen storage diseases-time to flip the outdated diagnostic apDocument4 pagesGlycogen storage diseases-time to flip the outdated diagnostic apAmber LatifNo ratings yet
- Carbohydrate Metabolism Catabolism Blok 7 2018Document136 pagesCarbohydrate Metabolism Catabolism Blok 7 2018N A Anggriani WulandariNo ratings yet
- Research Biochem G6PDHDocument5 pagesResearch Biochem G6PDHJohn Whel MararacNo ratings yet
- Carbohydrate MetabolismDocument56 pagesCarbohydrate MetabolismPradini SugihartoNo ratings yet
- S13.CHO .MetabolismDocument18 pagesS13.CHO .MetabolismGhea Jovita SinagaNo ratings yet
- In Vitro Stability and in Vivo Pharmacokinetic Studies of A Model Opioid Peptide, H-Tyr - Ala-Gly-Phe - Leu-OH (DADLE), and Its Cyclic ProdrugsDocument9 pagesIn Vitro Stability and in Vivo Pharmacokinetic Studies of A Model Opioid Peptide, H-Tyr - Ala-Gly-Phe - Leu-OH (DADLE), and Its Cyclic ProdrugskhurramwmNo ratings yet
- Substrates&EnzymesDocument15 pagesSubstrates&EnzymesTanyaTouchéNo ratings yet
- Glycolysis ASAS 4104Document8 pagesGlycolysis ASAS 4104AlbanMugotiNo ratings yet
- 371Document10 pages371kevincan100% (1)
- Non Energy Role of CarbohydratesDocument34 pagesNon Energy Role of CarbohydratesDr. M. Prasad NaiduNo ratings yet
- Lecture 03. Carbohydrate Metabolism. Common Pathway, Regulation and PathologyDocument80 pagesLecture 03. Carbohydrate Metabolism. Common Pathway, Regulation and PathologyВіталій Михайлович НечипорукNo ratings yet
- Methods For Preclinical Evaluation of Bioactive Natural ProductsFrom EverandMethods For Preclinical Evaluation of Bioactive Natural ProductsNo ratings yet
- Figure 1_ Geographic distribution of parasites and SNPs. _ Nature GeneticsDocument1 pageFigure 1_ Geographic distribution of parasites and SNPs. _ Nature GeneticssethawudNo ratings yet
- Forecasting for the Long-Term and the Short-Term Quiz _ 2Document4 pagesForecasting for the Long-Term and the Short-Term Quiz _ 2sethawudNo ratings yet
- 2008 The Structure of A Chondroitin Sulfate-Binding Domain Important in Placental MalariaDocument6 pages2008 The Structure of A Chondroitin Sulfate-Binding Domain Important in Placental MalariasethawudNo ratings yet
- 2009 Simultaneous Transcription of Duplicated Var2csa Gene Copies in Individual Plasmodium Falciparum ParasitesDocument16 pages2009 Simultaneous Transcription of Duplicated Var2csa Gene Copies in Individual Plasmodium Falciparum ParasitessethawudNo ratings yet
- 2008 VAR2CSA Domains Expressed in Escherichia Coli Induce Cross-Reactive Antibodies To Native ProteinDocument5 pages2008 VAR2CSA Domains Expressed in Escherichia Coli Induce Cross-Reactive Antibodies To Native ProteinsethawudNo ratings yet
- 2007 Effects of Sex, Parity, and Sequence Variation On Seroreactivity To Candidate Pregnancy Malaria Vaccine AntigensDocument10 pages2007 Effects of Sex, Parity, and Sequence Variation On Seroreactivity To Candidate Pregnancy Malaria Vaccine AntigenssethawudNo ratings yet
- Michigan's Revised Stay-At-Home OrderDocument10 pagesMichigan's Revised Stay-At-Home OrderMLive.com67% (3)
- ARDS Care Respiratory Care Plan PDFDocument2 pagesARDS Care Respiratory Care Plan PDFeric parlNo ratings yet
- Characteristics of Neonate (Part 1)Document52 pagesCharacteristics of Neonate (Part 1)Additi SatyalNo ratings yet
- Mono 100 CDocument527 pagesMono 100 CLe KhaiNo ratings yet
- Field Visit Report To Vrindavan Botanical Garden and Lumle Regional Agricultural Research StationDocument43 pagesField Visit Report To Vrindavan Botanical Garden and Lumle Regional Agricultural Research StationAmresh kumar Shah100% (1)
- Finally Complete Thssis Nuuradiin Last DraffDocument25 pagesFinally Complete Thssis Nuuradiin Last DraffAbdi KhadarNo ratings yet
- Scope of Nle919Document305 pagesScope of Nle919ericNo ratings yet
- Georges Cuvier's Theory of Correlation of PartsDocument5 pagesGeorges Cuvier's Theory of Correlation of PartsSoc SaballaNo ratings yet
- Gordon & PADocument12 pagesGordon & PAKristine AlejandroNo ratings yet
- Ards in Traumatic Brain InjuryDocument33 pagesArds in Traumatic Brain InjuryPiyush GirdharNo ratings yet
- Lecture Notes On Medical Parasitology (Introduction) By: Gessessew Bugssa, Mekelle UniversityDocument24 pagesLecture Notes On Medical Parasitology (Introduction) By: Gessessew Bugssa, Mekelle UniversityGessessew Bugssa100% (10)
- GastritisDocument3 pagesGastritisaliceNo ratings yet
- Immature GranulocytesDocument10 pagesImmature Granulocytespieterinpretoria391No ratings yet
- Non Spore Forming, Nonbranching Catalase Positive BacilliDocument5 pagesNon Spore Forming, Nonbranching Catalase Positive BacilliFaithNo ratings yet
- Magnesium Sulphate in The EmergencyDocument12 pagesMagnesium Sulphate in The EmergencyNur AzizaNo ratings yet
- OtosclerosisDocument38 pagesOtosclerosisSushmita Kaldan100% (1)
- Cell TransportDocument7 pagesCell TransportMillenNo ratings yet
- Jurnal Retardasi MentalDocument10 pagesJurnal Retardasi MentalfarahNo ratings yet
- WASH Practitioners Manual - Accessibility of Public & School Toilet For Indians With DisabilitiesDocument76 pagesWASH Practitioners Manual - Accessibility of Public & School Toilet For Indians With DisabilitiesVaishnavi JayakumarNo ratings yet
- Study of Various Constitutions in Females From MurphyDocument5 pagesStudy of Various Constitutions in Females From MurphyShubhanshi BhasinNo ratings yet
- Mi RNADocument14 pagesMi RNATeoMartinNo ratings yet
- Yr 7 Vocab ReproductionDocument2 pagesYr 7 Vocab ReproductionfghfjffgfgjfgfjNo ratings yet
- Bpdy Weight and Vitamin DDocument14 pagesBpdy Weight and Vitamin DMarijosse NavarroNo ratings yet
- David Grove Clean Questions For AddictionDocument12 pagesDavid Grove Clean Questions For AddictionAnonymous 2a1YI92k9L100% (6)
- UNIT III A in Microbiology and ParasitologyDocument8 pagesUNIT III A in Microbiology and ParasitologyBrixValdrizNo ratings yet
- Butea Superba Sci - The Use of Butea Superba (Roxb.) Compared To Sildenafil For Treating Erectile DysfunctionDocument5 pagesButea Superba Sci - The Use of Butea Superba (Roxb.) Compared To Sildenafil For Treating Erectile DysfunctionyunusNo ratings yet
- Psychoanalysis of Gifted Minds PSYDocument12 pagesPsychoanalysis of Gifted Minds PSYVirat SinghNo ratings yet
- Kanner AutismDocument5 pagesKanner AutismNinelys CodNo ratings yet
- Ending Discrimination Against People With Mental and Substance Use Disorders: The Evidence For Stigma ChangeDocument171 pagesEnding Discrimination Against People With Mental and Substance Use Disorders: The Evidence For Stigma ChangeJose Fernandez MNo ratings yet