Download as pdf or txt
You might also like
- ES.0.06.0021 - Project DossierDocument31 pagesES.0.06.0021 - Project DossierSaravanan Varadarajan100% (1)
- Estimation of PKaDocument3 pagesEstimation of PKaLiliana Andrea Pacheco Miranda100% (1)
- Solvent Effects On The Singlet - Triplet Equilibrium and Reactivity of A Ground Triplet State Arylalkyl CarbeneDocument4 pagesSolvent Effects On The Singlet - Triplet Equilibrium and Reactivity of A Ground Triplet State Arylalkyl CarbeneSergioSilvaNo ratings yet
- 8a Miller1995Document2 pages8a Miller1995Tu NaruNo ratings yet
- EJOC1999Document4 pagesEJOC1999ezhibobrikNo ratings yet
- A Covalently Bound Dimeric Derivative of Pyrochlorophyllide A Possible Model For Reaction Center Chlorophyll'Document3 pagesA Covalently Bound Dimeric Derivative of Pyrochlorophyllide A Possible Model For Reaction Center Chlorophyll'Mohammed ZiyadNo ratings yet
- Wen Fu1996Document6 pagesWen Fu1996scarlett salasNo ratings yet
- Trost 2004Document5 pagesTrost 2004uzacccNo ratings yet
- Poly EneDocument3 pagesPoly EneMohammed TarekNo ratings yet
- Reaccion de Eliminacion E y ZDocument5 pagesReaccion de Eliminacion E y ZKarelis GutierrezNo ratings yet
- Docx1 Lab ReportDocument5 pagesDocx1 Lab ReportM Zeeshan aliNo ratings yet
- Teresita Maldonando, Eduardo Martínez-González, Carlos FrontanaDocument1 pageTeresita Maldonando, Eduardo Martínez-González, Carlos FrontanaartedlcNo ratings yet
- Role of Solvent Reorganization Dynamics in Electron-Transfer Processes. Anomalous Kinetic Behavior in Alcohol SolventsDocument8 pagesRole of Solvent Reorganization Dynamics in Electron-Transfer Processes. Anomalous Kinetic Behavior in Alcohol Solventsenaveen2005No ratings yet
- Theoretical Study On The Mechanism of Robinson's Synthesis of TropinoneDocument4 pagesTheoretical Study On The Mechanism of Robinson's Synthesis of TropinoneDr-Muhammad Imran TousifNo ratings yet
- Tructural Hemistry Ommunications: DFT Based Characterization of Some Heterocyclic Compounds and Their Biological StudiesDocument10 pagesTructural Hemistry Ommunications: DFT Based Characterization of Some Heterocyclic Compounds and Their Biological StudiesAlex-Mihai CiubaraNo ratings yet
- Organic Chemistry 2021Document26 pagesOrganic Chemistry 2021xapodi8776No ratings yet
- Effect of β-Cyclodextrin on the Thermal Cis Trans Isomerization of AzobenzenesDocument6 pagesEffect of β-Cyclodextrin on the Thermal Cis Trans Isomerization of AzobenzenesnataliaNo ratings yet
- NMR N M R: Uclear Agnetic EsonanceDocument33 pagesNMR N M R: Uclear Agnetic EsonanceCarolina AlpucheNo ratings yet
- Martinezsaavedra 2014Document14 pagesMartinezsaavedra 2014snehasis banerjeeNo ratings yet
- Sulfonyl Esters. 2. CS Cleavage in Some Substitution Reactions of NitrobenzenesulfonatesDocument6 pagesSulfonyl Esters. 2. CS Cleavage in Some Substitution Reactions of NitrobenzenesulfonatesNik NorjumaNo ratings yet
- Unidad 3 - Parte 2 - Reacciones de Complejos de CoordinaciónDocument42 pagesUnidad 3 - Parte 2 - Reacciones de Complejos de CoordinaciónLUIS CARLOS ROMERO ZAPATA100% (1)
- NMRDocument16 pagesNMRChandni SahaNo ratings yet
- Diastereoselective Corey Chaykovsky 9 Epoxymethylation of Cinchona Alkaloids: Access To Chiral Scaffolds With Diverse FunctionalitiesDocument10 pagesDiastereoselective Corey Chaykovsky 9 Epoxymethylation of Cinchona Alkaloids: Access To Chiral Scaffolds With Diverse FunctionalitiesAna BrunoNo ratings yet
- Oxime DecompositionDocument6 pagesOxime DecompositionAmanda CamillaNo ratings yet
- Aromatic-Aromatic Interactions Free Energy Profiles For The Benzene Dimer in Water Chloroform and Liquid BenzeneDocument7 pagesAromatic-Aromatic Interactions Free Energy Profiles For The Benzene Dimer in Water Chloroform and Liquid BenzeneEsteban ArayaNo ratings yet
- Billones DomainDocument62 pagesBillones DomainZabrina Tan CuaNo ratings yet
- Key For Dec 2022 - Jan 2023 PaperDocument13 pagesKey For Dec 2022 - Jan 2023 PaperDivya ReddyNo ratings yet
- Lab Report - Thermodynamics and Kinetics of A Substitution Reaction of A Metal ComplexDocument16 pagesLab Report - Thermodynamics and Kinetics of A Substitution Reaction of A Metal ComplexValerie MangasarNo ratings yet
- Tugas Kelompok 3 - Peto Syarif - 20034071Document4 pagesTugas Kelompok 3 - Peto Syarif - 20034071peto syarifNo ratings yet
- Dominikus - Jurnal Reaksi Perisiklik 2Document3 pagesDominikus - Jurnal Reaksi Perisiklik 2Ekin Dwi ArifNo ratings yet
- Notes Lecture 1 Conformational AnalysisDocument18 pagesNotes Lecture 1 Conformational AnalysisDianing Wismarani Putri100% (1)
- Jacs AsapDocument2 pagesJacs AsapMohamadMostafaviNo ratings yet
- Organic Chemistry III Laboratory: NMR Verification of Diastereoselective Reduction of Substituted CyclohexanonesDocument4 pagesOrganic Chemistry III Laboratory: NMR Verification of Diastereoselective Reduction of Substituted Cyclohexanonesungu_sakuraNo ratings yet
- Effect of Cation On Room Temperature Ionic LiquidsDocument6 pagesEffect of Cation On Room Temperature Ionic LiquidsGRangarajanNo ratings yet
- Azeotropic ConditionsDocument7 pagesAzeotropic Conditionsmike-campbell-7340No ratings yet
- 1 s2.0 0584853971800387 Main PDFDocument7 pages1 s2.0 0584853971800387 Main PDFShreetama BhattacharyaNo ratings yet
- Zaky Al-FatonyDocument16 pagesZaky Al-FatonyZakyAlFatonyNo ratings yet
- Disrupt PolymersDocument2 pagesDisrupt PolymersBryan396No ratings yet
- 4811Document7 pages4811zosuaNo ratings yet
- Chem 203 Synthesis FFRDocument6 pagesChem 203 Synthesis FFRapi-261090898No ratings yet
- tmpE9D7 TMPDocument9 pagestmpE9D7 TMPFrontiersNo ratings yet
- Kin 20149Document6 pagesKin 20149Bruna ButkeNo ratings yet
- Spino1998 Journal of Organic Chemistry, 1998, Vol. 63, # 15, P. 5283 - 5287Document5 pagesSpino1998 Journal of Organic Chemistry, 1998, Vol. 63, # 15, P. 5283 - 5287SpazzaturaNo ratings yet
- J. Patrick A. Fairclough Et Al - Chain Length Dependence of The Mean Ðeld Temperature in Poly (Oxyethylene) - Poly (Oxybutylene) Diblock CopolymersDocument3 pagesJ. Patrick A. Fairclough Et Al - Chain Length Dependence of The Mean Ðeld Temperature in Poly (Oxyethylene) - Poly (Oxybutylene) Diblock CopolymersDremHpNo ratings yet
- Morcillo Et Al. (1987)Document17 pagesMorcillo Et Al. (1987)CLINT NELSONNo ratings yet
- Esch Et AlDocument24 pagesEsch Et AlFlopcornNo ratings yet
- Qafoku 2006Document15 pagesQafoku 2006Jaime Jaramillo GutierrezNo ratings yet
- Highly Enantioselective (4 + 2) Cycloaddition Reactions Catalyzed by A Chiral N-Methyl-oxazaborolidinium CationDocument3 pagesHighly Enantioselective (4 + 2) Cycloaddition Reactions Catalyzed by A Chiral N-Methyl-oxazaborolidinium Cationanuar_caldonNo ratings yet
- Ricardo Ugarte, Carlos Bustos, Ignacio Moreno-VillosladaDocument7 pagesRicardo Ugarte, Carlos Bustos, Ignacio Moreno-VillosladaSOCKYNo ratings yet
- Computation 02 00102Document10 pagesComputation 02 00102cmyk69No ratings yet
- Meta-Conjugation and Excited-State Coupling in Phenylacetylene DendrimersDocument2 pagesMeta-Conjugation and Excited-State Coupling in Phenylacetylene DendrimersGlade680No ratings yet
- Ferric Thio Cyan AteDocument16 pagesFerric Thio Cyan AtePablo BernalNo ratings yet
- Synthesis and DFT Studies of Novel Aryloxymaleimides Via Nucleophilic Substitution of Tosyloxy GroupDocument5 pagesSynthesis and DFT Studies of Novel Aryloxymaleimides Via Nucleophilic Substitution of Tosyloxy GroupCINDY VANESSA RESTREPO BURGOSNo ratings yet
- Evolution of Structure and Reactivity in A Series of Iconic CarbenesDocument8 pagesEvolution of Structure and Reactivity in A Series of Iconic CarbenesDiogo DiasNo ratings yet
- Paper Isomerization Nitrito Complejos CoDocument7 pagesPaper Isomerization Nitrito Complejos CoJuan Gabriel FernándezNo ratings yet
- Organic 2019Document24 pagesOrganic 2019xapodi8776No ratings yet
- Angela Seidl and Hans-Jurgen Hinz - The Free Energy of DNA Supercoiling Is Enthalpy-DeterminedDocument5 pagesAngela Seidl and Hans-Jurgen Hinz - The Free Energy of DNA Supercoiling Is Enthalpy-DeterminedDopameNo ratings yet
- Informe - Titulacion Potenciometrica Del Acido FosforicoDocument10 pagesInforme - Titulacion Potenciometrica Del Acido FosforicoScarlet Jacqueline Salas CalvoNo ratings yet
- 7745 PDFDocument12 pages7745 PDFDiogomussumNo ratings yet
- Henry S. Rzepa and Nikola Sanderson - Aromaticity On The Edge of Chaos: An Ab Initio Study of The Bimodal Balance Between Aromatic and Non-Aromatic Structures For 10p-Dihetero (8) AnnulenesDocument4 pagesHenry S. Rzepa and Nikola Sanderson - Aromaticity On The Edge of Chaos: An Ab Initio Study of The Bimodal Balance Between Aromatic and Non-Aromatic Structures For 10p-Dihetero (8) AnnulenesCvgdnnnyNo ratings yet
- Computational Pharmaceutics: Application of Molecular Modeling in Drug DeliveryFrom EverandComputational Pharmaceutics: Application of Molecular Modeling in Drug DeliveryDefang OuyangNo ratings yet
- ლიბერალი, მაისი 2010Document52 pagesლიბერალი, მაისი 2010emediageNo ratings yet
- Fenvik, მარტი 2012Document7 pagesFenvik, მარტი 2012emediageNo ratings yet
- დიალოგი, ივნისი 2010Document76 pagesდიალოგი, ივნისი 2010emediageNo ratings yet
- დიალოგი, იანვარი 2012Document68 pagesდიალოგი, იანვარი 2012emediageNo ratings yet
- დიალოგი, ივნისი 2011Document76 pagesდიალოგი, ივნისი 2011emediageNo ratings yet
- Fenvik, მარტი 2012Document180 pagesFenvik, მარტი 2012emediageNo ratings yet
- Fenvik, მარტი 2012Document50 pagesFenvik, მარტი 2012emediageNo ratings yet
- Seventeen, Aug 2011Document5 pagesSeventeen, Aug 2011emediageNo ratings yet
- Fenvik, მარტი 2012Document180 pagesFenvik, მარტი 2012emediageNo ratings yet
- Seventeen, Oct 2011Document4 pagesSeventeen, Oct 2011emediageNo ratings yet
- Seventeen, Sep 2011Document6 pagesSeventeen, Sep 2011emediageNo ratings yet
- მირიანა, თებერვალი 2012Document100 pagesმირიანა, თებერვალი 2012emediageNo ratings yet
- You and Your Family, Oct 2011Document5 pagesYou and Your Family, Oct 2011emediageNo ratings yet
- Seventeen, Jul 2011Document4 pagesSeventeen, Jul 2011emediageNo ratings yet
- Seventeen, Jul 2011Document4 pagesSeventeen, Jul 2011emediageNo ratings yet
- Assessment Plan ShilohDocument2 pagesAssessment Plan Shilohapi-478596695No ratings yet
- Business Intelligence: ComponentsDocument5 pagesBusiness Intelligence: ComponentslaptoptabingNo ratings yet
- FK76M E LoresDocument18 pagesFK76M E LoresFRANCONo ratings yet
- CP15 Semester 4 PDFDocument33 pagesCP15 Semester 4 PDFmithunprayagNo ratings yet
- Wrca Culminating-Activity Las-1Document6 pagesWrca Culminating-Activity Las-1Janah Rose Villagomez Peñaflor100% (1)
- DELMIA Digital Manufacturing Portfolio: IBM Product Lifecycle ManagementDocument20 pagesDELMIA Digital Manufacturing Portfolio: IBM Product Lifecycle ManagementsrinivaschakriNo ratings yet
- Immaculate Conception CollegeDocument4 pagesImmaculate Conception CollegeKester VillagonzaloNo ratings yet
- Question Number One (20 Points (ADocument4 pagesQuestion Number One (20 Points (AabuahmadnoorNo ratings yet
- SN2700 Open Ethernet Switch: HighlightsDocument3 pagesSN2700 Open Ethernet Switch: HighlightsKhawar NehalNo ratings yet
- Ora Laboratory Manual: Section 4 Section 4Document78 pagesOra Laboratory Manual: Section 4 Section 4Joanna Mae Masigan DacanayNo ratings yet
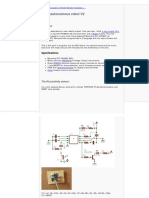
- How-To Build A Little Autonomous Robot With PIC 18F4520 PDFDocument22 pagesHow-To Build A Little Autonomous Robot With PIC 18F4520 PDFanselmoNo ratings yet
- EuPhO 2019 Theory ProblemsDocument1 pageEuPhO 2019 Theory ProblemsKolisetty SudhakarNo ratings yet
- ArticleDocument2 pagesArticleminiriniNo ratings yet
- Track Drainage - NSW - Very GoodDocument83 pagesTrack Drainage - NSW - Very GoodFebrian QodriNo ratings yet
- Kesantunan Berbahasa Dalam Masyarakat Osing Di Kabupaten BanyuwangiDocument18 pagesKesantunan Berbahasa Dalam Masyarakat Osing Di Kabupaten BanyuwangiBriliantoko Bagus WicaksonoNo ratings yet
- SmartSniff - Packet Sniffer - Capture TCP-IP Packets On Your Network AdapterDocument9 pagesSmartSniff - Packet Sniffer - Capture TCP-IP Packets On Your Network AdapterdrukovNo ratings yet
- Micromine LAB Manual-1Document12 pagesMicromine LAB Manual-1ercanpekNo ratings yet
- Measurement WorksheetDocument2 pagesMeasurement Worksheetgregory jimenezNo ratings yet
- 2019 Report WritingDocument9 pages2019 Report WritingRajaram PandaNo ratings yet
- Space Flight Mechanics AssignmentDocument2 pagesSpace Flight Mechanics AssignmentbooksforfunNo ratings yet
- Data Quality and CleaningDocument9 pagesData Quality and CleaningPadli SaedNo ratings yet
- Country N Code Continent Continent Income Grassociatioasia-Pacif Organisatifemale ParlDocument54 pagesCountry N Code Continent Continent Income Grassociatioasia-Pacif Organisatifemale ParllonelylokNo ratings yet
- P.5 Set 3 MTC Exam - Teacher - AcDocument11 pagesP.5 Set 3 MTC Exam - Teacher - AcSebuta HuzaimaNo ratings yet
- Comp Skills Isah 2Document6 pagesComp Skills Isah 2Baba HansNo ratings yet
- FLIR A325sc DatasheetDocument2 pagesFLIR A325sc Datasheetlegato13No ratings yet
- Urdu Model VerbsDocument8 pagesUrdu Model VerbsShabbir HuaasinNo ratings yet
- PG32 (1176)Document3 pagesPG32 (1176)anand.bharadwajNo ratings yet
- Digital Camra DesignDocument47 pagesDigital Camra DesignbabanpNo ratings yet