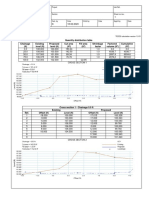
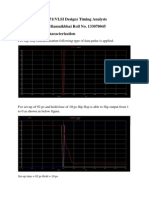
Download as pdf or txt
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5820)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (845)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Subunidad SigmaDocument5 pagesSubunidad SigmaGeorgina HernandezNo ratings yet
- Assembly of Recombinant Tfiid Reveals Differential Coactivator Requirements For Distinct Transcriptional ActivatorsDocument13 pagesAssembly of Recombinant Tfiid Reveals Differential Coactivator Requirements For Distinct Transcriptional ActivatorsGeorgina HernandezNo ratings yet
- A Transcription ReinitiationDocument5 pagesA Transcription ReinitiationGeorgina HernandezNo ratings yet
- Complement RecBCDDocument2 pagesComplement RecBCDGeorgina HernandezNo ratings yet
- © 1995 Nature Publishing GroupDocument8 pages© 1995 Nature Publishing GroupGeorgina HernandezNo ratings yet
- Supplementary Figures, Legends and Methods: Reca Transactivation of Pol VDocument9 pagesSupplementary Figures, Legends and Methods: Reca Transactivation of Pol VGeorgina HernandezNo ratings yet
- BypassDocument5 pagesBypassGeorgina HernandezNo ratings yet
- Prim As ADocument7 pagesPrim As AGeorgina HernandezNo ratings yet
- Nature Jan 18, 1996 379, 6562 ProquestDocument8 pagesNature Jan 18, 1996 379, 6562 ProquestGeorgina HernandezNo ratings yet
- AcknowledgementDocument7 pagesAcknowledgementElan Chezhian50% (2)
- Cut and Fill ExampleDocument3 pagesCut and Fill ExampleMallesh NenkatNo ratings yet
- Work Study and ErgonomicsDocument4 pagesWork Study and ErgonomicsVikas VarmaNo ratings yet
- Chapter 1Document8 pagesChapter 1Cj Lopez100% (2)
- Philosophy of Architecture Book-Of-AbstractsDocument70 pagesPhilosophy of Architecture Book-Of-AbstractsIdaHNo ratings yet
- Lesson PlanDocument4 pagesLesson PlanDieg Go0% (1)
- Vocation NotesDocument16 pagesVocation NotessankarjvNo ratings yet
- CMDB2.0.1 DevelopersReferenceGuideDocument386 pagesCMDB2.0.1 DevelopersReferenceGuidemarkiitotNo ratings yet
- Humastar 80Document4 pagesHumastar 80rizal_aspanNo ratings yet
- Highlights of The YearDocument3 pagesHighlights of The YearbhaveshlibNo ratings yet
- HR KPIsDocument151 pagesHR KPIstaapNo ratings yet
- 16 Bit Accumulator Using NAND and Logical Effort MethodDocument22 pages16 Bit Accumulator Using NAND and Logical Effort MethodJaydip FadaduNo ratings yet
- Keneuoe MohlakoanaDocument137 pagesKeneuoe MohlakoanaDurga BhavaniNo ratings yet
- Logical Correlation Analysis On Victim and Aggressor Pairs With The Same SenseDocument3 pagesLogical Correlation Analysis On Victim and Aggressor Pairs With The Same SenseSumanth VarmaNo ratings yet
- 2080 Um002 - en e PDFDocument270 pages2080 Um002 - en e PDFΔημητρηςΣαρακυρουNo ratings yet
- Social Structure of PakistanDocument3 pagesSocial Structure of PakistanFaizan ButtNo ratings yet
- 02 - Flaws in EuclidDocument32 pages02 - Flaws in EuclidRiemann SolivenNo ratings yet
- Science 7 Scientific Methods Week 1Document3 pagesScience 7 Scientific Methods Week 1Samina ManasNo ratings yet
- 411-5221-955.08.08 SGSN User GuideDocument622 pages411-5221-955.08.08 SGSN User Guideav8r_txNo ratings yet
- Published On Landscape ArchitectureDocument7 pagesPublished On Landscape ArchitectureShrila MitraNo ratings yet
- Science and Philosophy in Ancient IndiaDocument22 pagesScience and Philosophy in Ancient IndiaSobhan DasariNo ratings yet
- LiveReport 01 19Document46 pagesLiveReport 01 19rasnimNo ratings yet
- Human Flourishing-M. HeideggerDocument2 pagesHuman Flourishing-M. Heideggerpot pooot100% (2)
- Journalism 4Document8 pagesJournalism 4Lileth Fabio-oliverioNo ratings yet
- On A Bivariate PoissonDocument11 pagesOn A Bivariate PoissonAsif ThottathilNo ratings yet
- WWW Successcds Net Learn English Class 10 Important Questions The Thiefs Story CDocument27 pagesWWW Successcds Net Learn English Class 10 Important Questions The Thiefs Story CAnushka SenNo ratings yet
- Req Specification2017Document65 pagesReq Specification2017polooNo ratings yet
- SP Efsa 2009 EN-14 PDFDocument396 pagesSP Efsa 2009 EN-14 PDFtammineedi ramuNo ratings yet
- Chapter 1 Basic Properties of IntegersDocument40 pagesChapter 1 Basic Properties of IntegersZebider Ayenew AgezeNo ratings yet
- Laura Bohorquez ResumeDocument2 pagesLaura Bohorquez Resumeapi-268779952No ratings yet