Download as pptx, pdf, or txt
You might also like
- Oxford Handbook of Clinical Medicine 11eDocument93 pagesOxford Handbook of Clinical Medicine 11ethesagordey33% (3)
- Harrison's Principles of Internal Medicine 20th Edition (Vol.1 & Vol.2) 2018 PDFDocument866 pagesHarrison's Principles of Internal Medicine 20th Edition (Vol.1 & Vol.2) 2018 PDFJude Eric Dela Cruz75% (53)
- Statics - Centroid & Center of MassDocument8 pagesStatics - Centroid & Center of MassEuw Chaiwanont100% (1)
- Medical Physiology, Boron, Boulpaep 812Document13 pagesMedical Physiology, Boron, Boulpaep 812Kristijan Goldašić50% (2)
- Pocket Neurology by M. Brandon Westover - M.D. Ph.D. - David M Greer - Emily Choi - Karim M. Awad - M.D.Document30 pagesPocket Neurology by M. Brandon Westover - M.D. Ph.D. - David M Greer - Emily Choi - Karim M. Awad - M.D.aliss_wib9928100% (2)
- Hopkins Medicine Review Selected Topics in General and Internal MedicineDocument65 pagesHopkins Medicine Review Selected Topics in General and Internal Medicinetarun956519100% (1)
- Guided Notes - Production CurvesDocument7 pagesGuided Notes - Production CurvesyogurtkingyahooNo ratings yet
- (Ruth Ballweg, MPA, PA-C, Edward M. Sullivan, MS, (B-Ok - Xyz) PDFDocument939 pages(Ruth Ballweg, MPA, PA-C, Edward M. Sullivan, MS, (B-Ok - Xyz) PDFJim Velasquez100% (1)
- Harrisons Principles of Internal MedicineDocument9 pagesHarrisons Principles of Internal MedicineJennifer Gaspar14% (14)
- Harrison's Principles of Internal Medicine, Twentieth Edition (Vol.1 & Vol.2) - J. Larry JamesonDocument6 pagesHarrison's Principles of Internal Medicine, Twentieth Edition (Vol.1 & Vol.2) - J. Larry Jamesongilikixy33% (6)
- Harrisons Neurology in Clinical Medicine PDFDocument784 pagesHarrisons Neurology in Clinical Medicine PDFangga100% (21)
- Merrits 13thDocument2,669 pagesMerrits 13th星拉斐100% (3)
- Clinical Neurophysiology 3 e 2009Document915 pagesClinical Neurophysiology 3 e 2009Eldar Sulejmanovic100% (11)
- Fisiología Medica Raff PDFDocument801 pagesFisiología Medica Raff PDFYamelNo ratings yet
- @MedicalBooksStore 2014 Best of PDFDocument284 pages@MedicalBooksStore 2014 Best of PDFSaman SarKoNo ratings yet
- CV - David Gozal M.D. (USA)Document87 pagesCV - David Gozal M.D. (USA)MSKCNo ratings yet
- Endocrinology - Internal Medicine, Dr. A. Mowafy (2020-2021)Document172 pagesEndocrinology - Internal Medicine, Dr. A. Mowafy (2020-2021)Mohammed Risq100% (1)
- Internal MedicineDocument13 pagesInternal MedicineTianah davisNo ratings yet
- Machine Design Ebook1Document184 pagesMachine Design Ebook1Hew LetNo ratings yet
- NumerologyDocument24 pagesNumerologyphani60% (5)
- MRCP Part 2 2022 Dec Recall by SystemDocument5 pagesMRCP Part 2 2022 Dec Recall by SystemAlena JosephNo ratings yet
- Textbook of NeuroimagingDocument403 pagesTextbook of NeuroimagingcelooshNo ratings yet
- MRCPUK Sample Part 1 PDFDocument129 pagesMRCPUK Sample Part 1 PDFA.h.MuradNo ratings yet
- List of Clinical Medicine Books: Titles Authors Publisher YearDocument13 pagesList of Clinical Medicine Books: Titles Authors Publisher Yearwaqas_xsNo ratings yet
- Harrison's 17th EdDocument3 pagesHarrison's 17th EdShila Lupiyatama50% (2)
- NeurologyFrom EverandNeurologyStuart C. SealfonNo ratings yet
- Diabetes Problem Based Learning PDFDocument6 pagesDiabetes Problem Based Learning PDFIfantri Pramana100% (1)
- Basic CTDocument71 pagesBasic CTRapid Medicine100% (2)
- Cardiology Ebook Notes PDFDocument26 pagesCardiology Ebook Notes PDFsugisweNo ratings yet
- Core V - Cardiovascular CoreDocument35 pagesCore V - Cardiovascular CoreMatthew LeiNo ratings yet
- Harrison's™ Manual of Medicine - PDF Drive PDFDocument1 pageHarrison's™ Manual of Medicine - PDF Drive PDFEmma EshunNo ratings yet
- A Guide To MRCP (UK) and Specialty Certificate Examinations 2010Document6 pagesA Guide To MRCP (UK) and Specialty Certificate Examinations 2010rmaraj007100% (2)
- MRCP 2 Practice Questions Book.3Document166 pagesMRCP 2 Practice Questions Book.3iban100% (1)
- พี่พงษ์ติวcardioDocument79 pagesพี่พงษ์ติวcardioRapid Medicine67% (3)
- 55 Cases in NeurologyDocument419 pages55 Cases in NeurologykhawlahbintialazwarNo ratings yet
- Comprehensive Clinical Nephrology 4th Editionl PDFDocument4 pagesComprehensive Clinical Nephrology 4th Editionl PDFJenniferNo ratings yet
- Board Review Endocrinology A. ApiradeeDocument47 pagesBoard Review Endocrinology A. ApiradeePiyasak NaumnaNo ratings yet
- Neurology PearlsDocument18 pagesNeurology PearlsRjD100% (1)
- Portal Hypertension VIIDocument100 pagesPortal Hypertension VIIDiego V.G.No ratings yet
- Cardiology BookDocument401 pagesCardiology BookOana100% (5)
- @MBS MedicalBooksStore 2019 CardiovascularDocument273 pages@MBS MedicalBooksStore 2019 CardiovascularAhmad FitriawanNo ratings yet
- Peripheral Neuropathy Diff Diagnosis and Management AafpDocument6 pagesPeripheral Neuropathy Diff Diagnosis and Management Aafpgus_lions100% (1)
- Cardiology Crash CourseDocument6 pagesCardiology Crash CourseTashfiq HaiderNo ratings yet
- Valvular Heart Disease: Presented by DR Mirjana Milutinovic Professor, SJSMDocument69 pagesValvular Heart Disease: Presented by DR Mirjana Milutinovic Professor, SJSMAbanoub AwadallaNo ratings yet
- 1536106348Document144 pages1536106348Saman SarKoNo ratings yet
- EMP Enero 2011 Anticoagulacioìn en UrgenciaDocument20 pagesEMP Enero 2011 Anticoagulacioìn en UrgenciaNicolás HerreraNo ratings yet
- The Whole Enchilada PDFDocument602 pagesThe Whole Enchilada PDFMr XNo ratings yet
- Acid Base Workshop For OUWB HandoutDocument28 pagesAcid Base Workshop For OUWB HandoutJoel Topf100% (2)
- X-Ray CVSDocument67 pagesX-Ray CVSmdjohariNo ratings yet
- Self-Assessment: BOFs for MRCP(UK) and MRCP(I) Part IFrom EverandSelf-Assessment: BOFs for MRCP(UK) and MRCP(I) Part INo ratings yet
- Neurology Clerkship Study GuideDocument84 pagesNeurology Clerkship Study GuideHilary Steele100% (1)
- Adrian Covic, Mehmet Kanbay, Edgar V. Lerma (Eds.) - Dyslipidemias in Kidney Disease (2014, Springer-Verlag New York) PDFDocument325 pagesAdrian Covic, Mehmet Kanbay, Edgar V. Lerma (Eds.) - Dyslipidemias in Kidney Disease (2014, Springer-Verlag New York) PDFAndrada BararNo ratings yet
- Neurophysiology Made EasyDocument42 pagesNeurophysiology Made EasyNaotaka KohinataNo ratings yet
- The System: RespiDocument219 pagesThe System: Respilalaine22dale100% (1)
- Pharmacology of Pain: Proceedings of the First International Pharmacological Meeting, Stockholm, 22-25 August, 1961From EverandPharmacology of Pain: Proceedings of the First International Pharmacological Meeting, Stockholm, 22-25 August, 1961R. K. S. LimNo ratings yet
- Top Trials in Gastroenterology & HepatologyFrom EverandTop Trials in Gastroenterology & HepatologyRating: 4.5 out of 5 stars4.5/5 (7)
- Cardio-Hepatology: Connections Between Hepatic and Cardiovascular DiseaseFrom EverandCardio-Hepatology: Connections Between Hepatic and Cardiovascular DiseaseTatsunori TaniguchiNo ratings yet
- Interventional Neuroradiology A Complete Guide - 2020 EditionFrom EverandInterventional Neuroradiology A Complete Guide - 2020 EditionNo ratings yet
- Pharmacology of Antihypertensive DrugsFrom EverandPharmacology of Antihypertensive DrugsP.A. Van ZwietenRating: 3 out of 5 stars3/5 (1)
- FM Assignment Booster ChaiDocument26 pagesFM Assignment Booster ChaiAbin ShaNo ratings yet
- ISO 9001 Awareness AmaDocument51 pagesISO 9001 Awareness AmaHisar SimanjuntakNo ratings yet
- Utpaladeva's Lost Vivrti On The Ishwara Pratyabhijna Karika - Raffaele TorellaDocument12 pagesUtpaladeva's Lost Vivrti On The Ishwara Pratyabhijna Karika - Raffaele Torellaraffaeletorella100% (1)
- BIOLS102-UOB-Chapter 10Document8 pagesBIOLS102-UOB-Chapter 10Noor JanahiNo ratings yet
- Orangutan ListDocument5 pagesOrangutan ListRam ChandiranNo ratings yet
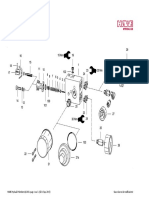
- Cutter-Marble - Bosch GDM13-34Document79 pagesCutter-Marble - Bosch GDM13-34tamilmanoharNo ratings yet
- Badenian Gastropods From The Collections of The Mureş County MuseumDocument26 pagesBadenian Gastropods From The Collections of The Mureş County MuseumLouisaMjjNo ratings yet
- A1 (2 3 4) F./... D 6905 Af/1Document3 pagesA1 (2 3 4) F./... D 6905 Af/1GERALD SIMONNo ratings yet
- JAMA 1990 Meshberger 1837 41 PDFDocument5 pagesJAMA 1990 Meshberger 1837 41 PDFNicholas TorreyNo ratings yet
- Etl Book PDFDocument2 pagesEtl Book PDFLaura0% (1)
- Notes On Jean Piaget DeweyDocument2 pagesNotes On Jean Piaget DeweyfadzillahNo ratings yet
- Evaluation of Trajectory Model For Differential Steering Using Numerical MethodDocument6 pagesEvaluation of Trajectory Model For Differential Steering Using Numerical MethodNaufal RachmatullahNo ratings yet
- The Duality of Human Nature in Oscar Wilde's The Importance of Being EarnestDocument28 pagesThe Duality of Human Nature in Oscar Wilde's The Importance of Being EarnestSowmya ShreeNo ratings yet
- S5750E-SI Series L2 Gigabits Dual Stack Intelligent Switch DatasheetDocument7 pagesS5750E-SI Series L2 Gigabits Dual Stack Intelligent Switch DatasheetThe LyricsNo ratings yet
- Shotgun StatisticsDocument1 pageShotgun Statistics-No ratings yet
- Matrices of Violence: A Post-Structural Feminist Rendering of Nawal El Saadawi's Woman at Point Zero and Lola Soneyin's The Secrets of Baba Segi's WivesDocument6 pagesMatrices of Violence: A Post-Structural Feminist Rendering of Nawal El Saadawi's Woman at Point Zero and Lola Soneyin's The Secrets of Baba Segi's WivesIJELS Research JournalNo ratings yet
- Welcome Back! B1 SVDocument4 pagesWelcome Back! B1 SVMagda StręciwilkNo ratings yet
- Weebly Homework Article Review Standard 2Document11 pagesWeebly Homework Article Review Standard 2api-289106854No ratings yet
- A&H Carrefour LayoutDocument1 pageA&H Carrefour LayoutAshraf EhabNo ratings yet
- Organisational BehaviourDocument9 pagesOrganisational BehaviourMuthu RamalakshmiNo ratings yet
- Scala AccesoriesDocument27 pagesScala AccesoriesZiggy BussyNo ratings yet
- Make Wire Transfers Through Online Banking From AnywhereDocument2 pagesMake Wire Transfers Through Online Banking From AnywhereDani PermanaNo ratings yet
- Fizika RadiologijaDocument9 pagesFizika RadiologijaBorislav TapavičkiNo ratings yet
- DKE344 BibDocument2 pagesDKE344 BibMohamad SleimanNo ratings yet
- 2021 HhhhhhhterqasdwqwDocument1 page2021 HhhhhhhterqasdwqwHassan KhanNo ratings yet
- A Practical Grammar of The Sanskrit LanguageDocument408 pagesA Practical Grammar of The Sanskrit LanguageungulataNo ratings yet