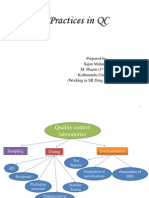
Download as ppt, pdf, or txt
You might also like
- M Test (Meridian Test) MukainoDocument11 pagesM Test (Meridian Test) MukainoVicente Acosta100% (1)
- Laboratory Quality/Management: A Workbook with an Eye on AccreditationFrom EverandLaboratory Quality/Management: A Workbook with an Eye on AccreditationRating: 5 out of 5 stars5/5 (1)
- Autism For DummiesDocument11 pagesAutism For Dummiesgocyndigo72yahoocomNo ratings yet
- Subiecte Olimpiadă Limba Engleză Clasa A 9aDocument3 pagesSubiecte Olimpiadă Limba Engleză Clasa A 9akallai annamaria100% (1)
- Chapter 1 - Part 1 Introduction To Quality AssuranceDocument25 pagesChapter 1 - Part 1 Introduction To Quality AssuranceLily ERc Peter100% (4)
- Skills ProgramDocument3 pagesSkills ProgramAbongile Phinyana100% (1)
- Differences Between #GoodManufacturingPractices (GMPS) and #GoodLaboratoryPractices (GLPS)Document55 pagesDifferences Between #GoodManufacturingPractices (GMPS) and #GoodLaboratoryPractices (GLPS)Silvia OrdazNo ratings yet
- Shayana GLP FinalDocument34 pagesShayana GLP FinalShayana GoraNo ratings yet
- Good Laboratory Practice: Presented To Presented byDocument39 pagesGood Laboratory Practice: Presented To Presented byaakash sahaNo ratings yet
- Good Laboratory Practice: Presented To Presented byDocument39 pagesGood Laboratory Practice: Presented To Presented byAbhitav TiwariNo ratings yet
- GLP - 2Document39 pagesGLP - 2AkshitaNo ratings yet
- All You Never Wanted To Know About GLP and GMPDocument55 pagesAll You Never Wanted To Know About GLP and GMPmuraliapNo ratings yet
- Checklist For Laboratory PDFDocument12 pagesChecklist For Laboratory PDFSharia Attar0% (1)
- Good Laboratory Practices (GLP)Document35 pagesGood Laboratory Practices (GLP)Sarah Tahir100% (1)
- GLP FinalDocument7 pagesGLP Finalaakash sahaNo ratings yet
- Chm561 Chapter 7 - Quality Systems in Chemical Laboratories-ReviewedDocument46 pagesChm561 Chapter 7 - Quality Systems in Chemical Laboratories-ReviewedLina LynxNo ratings yet
- Lec 5 (GLP)Document15 pagesLec 5 (GLP)sheikh muhammad mubashirNo ratings yet
- Good Laboratory Practices PDF by Dr. Abhishek PandeyDocument26 pagesGood Laboratory Practices PDF by Dr. Abhishek PandeyWidiyantoNo ratings yet
- Day 2 Talk 6 - The Difference Between GLP and GLPDocument38 pagesDay 2 Talk 6 - The Difference Between GLP and GLPAnonymous rUr4olUNo ratings yet
- Concept of Good Laboratory Practices (GLP)Document18 pagesConcept of Good Laboratory Practices (GLP)SRINIVASAN GnanasabapathyNo ratings yet
- Review Quality Control Approachesin Clinical LaboratoriesDocument6 pagesReview Quality Control Approachesin Clinical LaboratoriesengmfowziNo ratings yet
- Introduction Good Clinical Laboratory PracticeDocument32 pagesIntroduction Good Clinical Laboratory PracticeMuzeena MansoorNo ratings yet
- Introduction Good Clinical Laboratory PracticeDocument31 pagesIntroduction Good Clinical Laboratory PracticeNailen HurtadoNo ratings yet
- GLP or Good Laboratory PracticesDocument35 pagesGLP or Good Laboratory Practicesningsih rezekiNo ratings yet
- 11-Nature of TheClinicalLaboratoryDocument19 pages11-Nature of TheClinicalLaboratoryKathlyn Patricia RealNo ratings yet
- Good Clinical Laboratory Practice GCLPDocument36 pagesGood Clinical Laboratory Practice GCLPPatrick Kosgei100% (1)
- Quality Assurance in Micro LabDocument49 pagesQuality Assurance in Micro LabSadia AhmadNo ratings yet
- Unit IIIDocument46 pagesUnit IIIDIPALI TALELENo ratings yet
- BP606T Unit 4-6Document222 pagesBP606T Unit 4-6Gyampoh SolomonNo ratings yet
- GLP Interview PrepDocument4 pagesGLP Interview PreppoiuNo ratings yet
- Cofactor and CoenzymeDocument73 pagesCofactor and CoenzymeBIDINNo ratings yet
- Good Laboratory Practices PDFDocument111 pagesGood Laboratory Practices PDFClydeA.SardoncilloNo ratings yet
- GLP Vs GMP Vs GCPDocument40 pagesGLP Vs GMP Vs GCPsuhasdasNo ratings yet
- Good Laboratory PracticeDocument3 pagesGood Laboratory PracticeИвана СоковићNo ratings yet
- Duo IndustrialDocument6 pagesDuo IndustrialLeinner José Motta TrujilloNo ratings yet
- Notes 1 For 25-03 Good Laboratory Practices PDFDocument26 pagesNotes 1 For 25-03 Good Laboratory Practices PDFgaurav tiwari100% (1)
- UKCRC Self-Assessment QuestDocument9 pagesUKCRC Self-Assessment QuesthotwarezNo ratings yet
- Good Laboratory Practice (GLP)Document35 pagesGood Laboratory Practice (GLP)Ankita JatkarNo ratings yet
- ArticleDocument4 pagesArticleMario RodríguezNo ratings yet
- Good Laboratory PracticesDocument52 pagesGood Laboratory Practicesankita pathania100% (1)
- Good Laboratory PracticesDocument21 pagesGood Laboratory PracticesSajanMaharjan100% (1)
- Microbiology Audit Guidance EbookDocument23 pagesMicrobiology Audit Guidance EbookArmando Saldaña100% (1)
- Good Laboratory Practice (GLP) Requirements: An Overview: KeywordsDocument5 pagesGood Laboratory Practice (GLP) Requirements: An Overview: KeywordsMahmoud DomourNo ratings yet
- WHO TRS 961 Eng-80-100Document21 pagesWHO TRS 961 Eng-80-100Valery LopezNo ratings yet
- College of American Pathologists: Laboratory Accreditation ProgramDocument30 pagesCollege of American Pathologists: Laboratory Accreditation ProgramLeevan BautistaNo ratings yet
- General Information: Medical (Clinical) Lab - DefinitionDocument5 pagesGeneral Information: Medical (Clinical) Lab - DefinitionMihaela MileaNo ratings yet
- GLPDocument14 pagesGLPMusfira KhalidNo ratings yet
- Who GLPDocument41 pagesWho GLPvyugueNo ratings yet
- Good Laboratory PracticesDocument11 pagesGood Laboratory PracticesAkash GayanNo ratings yet
- Good Laboratory PracticesDocument16 pagesGood Laboratory PracticesMerlin DineshNo ratings yet
- Materi 2Document23 pagesMateri 2Murdifin SulawesiNo ratings yet
- Laboratory Policy and Procedural ManualsDocument31 pagesLaboratory Policy and Procedural ManualsMegbaru100% (2)
- 02good Laboratory Practice (GLP)Document26 pages02good Laboratory Practice (GLP)Ayalew DesyeNo ratings yet
- 1305 - Laboratory Quality Control Based On Risk ManagementDocument68 pages1305 - Laboratory Quality Control Based On Risk ManagementDayledaniel Sorveto100% (1)
- Pqa U3.2 GLPDocument71 pagesPqa U3.2 GLPAkankshaNo ratings yet
- POCT and PT by Group6bsmt3bDocument15 pagesPOCT and PT by Group6bsmt3bRuzzelle Mae ParasNo ratings yet
- Good Laboratory PracticeDocument28 pagesGood Laboratory PracticeKranthi KumarNo ratings yet
- AppNote - Pipette Quality ControlDocument5 pagesAppNote - Pipette Quality ControlfaizNo ratings yet
- CHAPTER 3 - Quality Aspects of Lab - 2014 - Clinical Biochemistry Metabolic andDocument6 pagesCHAPTER 3 - Quality Aspects of Lab - 2014 - Clinical Biochemistry Metabolic andnour achkarNo ratings yet
- Good Laboratory Practices (BPOM) EngDocument61 pagesGood Laboratory Practices (BPOM) EngWidiyantoNo ratings yet
- Good Laboratory Practices (GLPS)Document3 pagesGood Laboratory Practices (GLPS)muddassar nazarNo ratings yet
- Good Laboratory PracticesDocument24 pagesGood Laboratory PracticesGerald Limo Arap ChebiiNo ratings yet
- ICH GuidelinesDocument4 pagesICH Guidelinesaakash sahaNo ratings yet
- Cash Remittance PrecautionsDocument8 pagesCash Remittance Precautionsaakash sahaNo ratings yet
- Vivo Analyses of Experimental Animals (6,7)Document23 pagesVivo Analyses of Experimental Animals (6,7)aakash sahaNo ratings yet
- Specialist OfficerDocument2 pagesSpecialist Officeraakash sahaNo ratings yet
- Good Laboratory Practice: Presented To Presented byDocument39 pagesGood Laboratory Practice: Presented To Presented byaakash sahaNo ratings yet
- GLP FinalDocument7 pagesGLP Finalaakash sahaNo ratings yet
- A Century After Yehidar-Beshita (The Spanish U in Ethiopia) : Are We Prepared For The Next Pandemic?Document5 pagesA Century After Yehidar-Beshita (The Spanish U in Ethiopia) : Are We Prepared For The Next Pandemic?Yalew MekonnenNo ratings yet
- Sample BrochureDocument2 pagesSample BrochurealyssamciceroNo ratings yet
- Safety Training ProgramDocument43 pagesSafety Training ProgramBhavesh PatelNo ratings yet
- INDIA - NRHM Common Review Mission - Fifth ReportDocument252 pagesINDIA - NRHM Common Review Mission - Fifth ReportKaushik BanerjeeNo ratings yet
- Dr. Michael Tarver Emergency Suspension ComplaintDocument10 pagesDr. Michael Tarver Emergency Suspension ComplaintDentist The MenaceNo ratings yet
- "Take Off Patient/ Operan Jaga": Tugas Bahasa InggrisDocument3 pages"Take Off Patient/ Operan Jaga": Tugas Bahasa InggrisKurnia anggrainiNo ratings yet
- Effects of Massage and Acupressure On Relieving.10Document9 pagesEffects of Massage and Acupressure On Relieving.10Riska amaliaNo ratings yet
- RadiologijaDocument42 pagesRadiologijaAzra Bajrektarevic0% (1)
- Myanmar Health Assistant Association Vacancy Announcement (VA-057/2021 MHAA-HR)Document3 pagesMyanmar Health Assistant Association Vacancy Announcement (VA-057/2021 MHAA-HR)MNo ratings yet
- Health Infrastructure and Economic Development in IndiaDocument9 pagesHealth Infrastructure and Economic Development in IndiaAkanksha PalNo ratings yet
- 08 OHS Journal Aug 2012Document60 pages08 OHS Journal Aug 2012NazirAhmadBashiriNo ratings yet
- MBT and TraumaDocument19 pagesMBT and Traumassanagav0% (1)
- Medical CertificateDocument1 pageMedical CertificateMulpuri Vijaya KumarNo ratings yet
- Evolution of Bhramari Pranayam in The Management of Menopausal SyndromeDocument7 pagesEvolution of Bhramari Pranayam in The Management of Menopausal SyndromeDr Kirti BhatiNo ratings yet
- Public Perception of Mental Health in Iraq: Research Open AccessDocument11 pagesPublic Perception of Mental Health in Iraq: Research Open AccessSri HariNo ratings yet
- OET Test 5 Listening Answers - Part A and BDocument9 pagesOET Test 5 Listening Answers - Part A and Bjeet meharNo ratings yet
- The Asbestos Institute: Contractor / SupervisorDocument4 pagesThe Asbestos Institute: Contractor / Supervisordaniellewisat7387No ratings yet
- Uterine Sub InvolutionDocument1 pageUterine Sub Involutionkrystle0875% (4)
- Nurse Practitioner and Clinical Nurse Specialist Competencies For Older Adult CareDocument36 pagesNurse Practitioner and Clinical Nurse Specialist Competencies For Older Adult Carejosephabram051590No ratings yet
- Stigma, Prejudice, Discrimination and HealthDocument16 pagesStigma, Prejudice, Discrimination and Healthnicoleta5aldea100% (1)
- Activity 1 - CPHDocument2 pagesActivity 1 - CPHMA. ANDREA NICOLE DURANNo ratings yet
- Medical Reimbursement AptranscoDocument13 pagesMedical Reimbursement AptranscopenusilaNo ratings yet
- CASR Part 67 Ed. 1 Amdt 0 - Medical Standard and CertificationDocument43 pagesCASR Part 67 Ed. 1 Amdt 0 - Medical Standard and CertificationR SetiawanNo ratings yet
- Rincian Biaya Perawatan: Rsud SekayuDocument6 pagesRincian Biaya Perawatan: Rsud SekayuMarni EllyzaNo ratings yet
- Treatment in Epileptic Encephalopathy With ESES and Landau-Kleffner SyndromeDocument6 pagesTreatment in Epileptic Encephalopathy With ESES and Landau-Kleffner SyndromeDaniela TapiaNo ratings yet
- Nestle India & Its Resililent SpiritDocument15 pagesNestle India & Its Resililent SpiritPrerna RathiNo ratings yet