Download as ppt, pdf, or txt
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5825)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (903)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (541)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (823)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Xeloda Tablets Description:: BioactivationDocument19 pagesXeloda Tablets Description:: BioactivationDrogoman MarianNo ratings yet
- Medically Compromised PatientsDocument21 pagesMedically Compromised PatientsDyah Wulan RamadhaniNo ratings yet
- 13 Pulmonary EmbolismDocument45 pages13 Pulmonary EmbolismMiky DinuNo ratings yet
- Theophylline.: BNF DrugsDocument2 pagesTheophylline.: BNF Drugsgege0% (1)
- THROMBOPHLEBITISDocument25 pagesTHROMBOPHLEBITISCamille Maluenda - Tan100% (2)
- DaktarinDocument3 pagesDaktarinleweeNo ratings yet
- Wa ReduceDocument455 pagesWa ReduceMuhammad Amrie100% (1)
- 2020 ESC Guidelines For The Management of Acute Coronary Syndromes in Patients Presenting Without Persistent ST-segment (25-34)Document10 pages2020 ESC Guidelines For The Management of Acute Coronary Syndromes in Patients Presenting Without Persistent ST-segment (25-34)Sheila Setiawati TanzilNo ratings yet
- Anticoagulant PDFDocument1 pageAnticoagulant PDFAnonymous zmPIy39No ratings yet
- The Drug That Cracked Covid by Michael CapuzzoDocument15 pagesThe Drug That Cracked Covid by Michael CapuzzoAll News Pipeline0% (1)
- 2019/2020 REVISION TOOL: Short Guides For Key Therapeutic AreasDocument68 pages2019/2020 REVISION TOOL: Short Guides For Key Therapeutic AreasEsraa AbdelfattahNo ratings yet
- Treatment Considerations, and Nursing Implications: Cardiovascular Disease and HIV: PathophysiologyDocument12 pagesTreatment Considerations, and Nursing Implications: Cardiovascular Disease and HIV: PathophysiologyRicardo Costa da SilvaNo ratings yet
- Blood Clot Symptoms To KnowDocument6 pagesBlood Clot Symptoms To KnowRaprnaNo ratings yet
- Bader Madoukh, Ayman Battisha, Henry Ukwu, Mohammed Al-Sadawi, Shakil ShaikhDocument4 pagesBader Madoukh, Ayman Battisha, Henry Ukwu, Mohammed Al-Sadawi, Shakil ShaikhputriayuratnasariNo ratings yet
- ELIQUIS HCP Electronic Combo Dosing Guide - Desktop PDFDocument29 pagesELIQUIS HCP Electronic Combo Dosing Guide - Desktop PDFAndreea Livia DumitrescuNo ratings yet
- L2-SURG-Hemostasis, Surgical Bleeding, - Transfusion (Aug2521)Document8 pagesL2-SURG-Hemostasis, Surgical Bleeding, - Transfusion (Aug2521)Marc Lyndon CafinoNo ratings yet
- Newer Oral Anticoagulant: DR Shivaom Chaurasia Resident Internal MedicineDocument57 pagesNewer Oral Anticoagulant: DR Shivaom Chaurasia Resident Internal MedicineMuhammad Reza FirdausNo ratings yet
- Haem-Heparin Prescribing Form 725 CUH 20211123Document2 pagesHaem-Heparin Prescribing Form 725 CUH 20211123Huzaifa SiddiquiNo ratings yet
- Principles of Antiplatelet, Anticoagulant and Fibrinolytic TherapiesDocument50 pagesPrinciples of Antiplatelet, Anticoagulant and Fibrinolytic TherapiesAbby LiewNo ratings yet
- Guidelines For Anticoagulation Using WarfarinDocument23 pagesGuidelines For Anticoagulation Using WarfarinMrinmayeeDeshmukhNo ratings yet
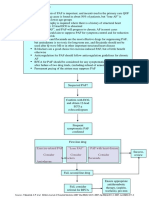
- Pathway DIAGNOSIS OF PAF FTRDocument1 pagePathway DIAGNOSIS OF PAF FTRYoan Eka Putra PalilingNo ratings yet
- Intracardiac Echocardiography2Document12 pagesIntracardiac Echocardiography2Salah AttaNo ratings yet
- Herb-Drug Interactions A Review and Study Based On Assessment of Clinical Case Reports in Literature Ph07033Document11 pagesHerb-Drug Interactions A Review and Study Based On Assessment of Clinical Case Reports in Literature Ph07033elokNo ratings yet
- Blood BankingDocument5 pagesBlood BankingTel LyNo ratings yet
- File Number 1 1-1000 General InstructionsDocument80 pagesFile Number 1 1-1000 General InstructionsDaniel RstomNo ratings yet
- Anti EmeticsDocument112 pagesAnti Emeticsjoel david knda mjNo ratings yet
- General Indications: AnticoagulantsDocument15 pagesGeneral Indications: AnticoagulantswahidNo ratings yet
- Apixaban: A Clinical Pharmacokinetic and Pharmacodynamic ReviewDocument15 pagesApixaban: A Clinical Pharmacokinetic and Pharmacodynamic ReviewNurul Kamilah SadliNo ratings yet
- ( ) هام للغايةHERB DRUG-INTERACTION-CHARTDocument5 pages( ) هام للغايةHERB DRUG-INTERACTION-CHARTALNAKINo ratings yet
- Book ReviewsDocument7 pagesBook Reviewsrahman10191871No ratings yet