Download as ppt, pdf, or txt
You might also like
- Case Presentation: Endometrial CancerDocument17 pagesCase Presentation: Endometrial Cancerxilefsemaj100% (4)
- DR Henry G Bieler Lines of Defense Against DiseaseDocument6 pagesDR Henry G Bieler Lines of Defense Against DiseasebonifacesilveiraNo ratings yet
- ESSENTIALS OF HUMAN EMBRYOLOGY by Prof NGASSAPADocument315 pagesESSENTIALS OF HUMAN EMBRYOLOGY by Prof NGASSAPAJumbe Mohamed100% (3)
- 209-Hematology Review - Case StudiesDocument129 pages209-Hematology Review - Case StudiesKhalid Khalidi100% (2)
- AnemiaDocument41 pagesAnemiaBang FadNo ratings yet
- Ancient Primer For Practical Godhead: The Deer Exercise For MenDocument25 pagesAncient Primer For Practical Godhead: The Deer Exercise For MenAnirudh100% (4)
- 5 - Anemias Associated With Abnormal Heme SynthesisDocument24 pages5 - Anemias Associated With Abnormal Heme SynthesisSara BakerNo ratings yet
- Clinical Hematological: Assist Prof. Dr. Mudhir S. ShekhaDocument31 pagesClinical Hematological: Assist Prof. Dr. Mudhir S. ShekhaAhmed. Masud.OthmanNo ratings yet
- MISDC AnaemiaDocument33 pagesMISDC AnaemiawanyNo ratings yet
- Primary HemochromatosisDocument36 pagesPrimary HemochromatosisKaish DahiyaNo ratings yet
- Haemolytic Anaemia: DR Nurul Fahmiza Tumiran P109273Document55 pagesHaemolytic Anaemia: DR Nurul Fahmiza Tumiran P109273Nurul FahmizaNo ratings yet
- Approach To A Patient With Anaemia.Document35 pagesApproach To A Patient With Anaemia.apule geraldhumbleNo ratings yet
- Classification and IDADocument32 pagesClassification and IDAdrshivukumarkpNo ratings yet
- Anemia Umam FazlurrahmanDocument44 pagesAnemia Umam Fazlurrahmanumam fazlurrahmanNo ratings yet
- Approached in Diagnosis of AnemiaDocument41 pagesApproached in Diagnosis of AnemiaAlqodri SetiawanNo ratings yet
- Red Cell DisordersDocument60 pagesRed Cell DisordersASMANI MATETENo ratings yet
- GIT Cirrhosis Liver in ChildrenDocument37 pagesGIT Cirrhosis Liver in ChildrenDr.P.NatarajanNo ratings yet
- 13.2 AnemiaDocument75 pages13.2 AnemiaMohammad AlrefaiNo ratings yet
- Approach To AnemiaDocument4 pagesApproach To AnemiapNo ratings yet
- Pancytopenia and Aplastic Anemia OkDocument33 pagesPancytopenia and Aplastic Anemia OkIrina MoldovanNo ratings yet
- Chapter 3 NotesDocument10 pagesChapter 3 Notesmjamie12345No ratings yet
- Haemolytic Anaemias 1Document41 pagesHaemolytic Anaemias 1mulengamordecai92No ratings yet
- AnemiaDocument108 pagesAnemiaAaron GarciaNo ratings yet
- 15 Sideroblastic AnemiaDocument24 pages15 Sideroblastic AnemiaKelvinTMaikanaNo ratings yet
- Haemolytic AnaemiasDocument71 pagesHaemolytic AnaemiasChipegoNo ratings yet
- Anemia: Reporter: FM R1 余明謙 Supervisor: Dr 陳信儒Document50 pagesAnemia: Reporter: FM R1 余明謙 Supervisor: Dr 陳信儒余明謙No ratings yet
- Chapter 2Document43 pagesChapter 2Puvaneswary SegharanNo ratings yet
- Diagnostic Approach To Anemia: Archana M Agarwal, M.DDocument46 pagesDiagnostic Approach To Anemia: Archana M Agarwal, M.DHasan AbdulhalemNo ratings yet
- Anaemia by Haider AliDocument38 pagesAnaemia by Haider AliAbdul SamadNo ratings yet
- Wilson's Disease: Dr. Imtiyaz Hashim PGR Mu-Iv (B)Document14 pagesWilson's Disease: Dr. Imtiyaz Hashim PGR Mu-Iv (B)امتیاز ہاشم بزنجوNo ratings yet
- Thalassemia 200510173444Document31 pagesThalassemia 200510173444sharifiNo ratings yet
- Anemias&Leukemias 1Document80 pagesAnemias&Leukemias 1Fallen AngelNo ratings yet
- Wisons DiseaseDocument8 pagesWisons DiseaseRani KhandelwalNo ratings yet
- HematuriaDocument42 pagesHematuriaAhmad SobihNo ratings yet
- Hematologic Disorders - CumulativeDocument28 pagesHematologic Disorders - CumulativeThesmith Fam100% (1)
- Baldwin Tub Ulo InterstitialDocument36 pagesBaldwin Tub Ulo InterstitialdrryanalwynNo ratings yet
- Dr. THOMAS KINGSLEY MD Professor of Medicine TVMCDocument48 pagesDr. THOMAS KINGSLEY MD Professor of Medicine TVMCDr THOMAS KINGSLEY MDNo ratings yet
- Rta Final Year 23.2.23Document36 pagesRta Final Year 23.2.23S.ayesh HasanNo ratings yet
- Bloodkb 160720181259Document129 pagesBloodkb 160720181259Jeena RajNo ratings yet
- 4) Inherited Hemolytic Anemia - Intraerythocytic With Defects of Haemoglobin Synthesis. Hemoglobinopathies and Thalassemia.Document2 pages4) Inherited Hemolytic Anemia - Intraerythocytic With Defects of Haemoglobin Synthesis. Hemoglobinopathies and Thalassemia.Lefteris ChatzidoukasNo ratings yet
- 1113 - PSBIM Review 2017 Hematology Lecture - IvyDocument253 pages1113 - PSBIM Review 2017 Hematology Lecture - IvyRye CalderonNo ratings yet
- Red Blood Cells DisordersDocument70 pagesRed Blood Cells Disordersluna zeidNo ratings yet
- Anemias ChartDocument14 pagesAnemias ChartM Patel100% (2)
- Presentation 5Document48 pagesPresentation 55xqqq8mx6pNo ratings yet
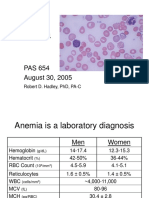
- Anemia Is A Laboratory Diagnosis: Men WomenDocument21 pagesAnemia Is A Laboratory Diagnosis: Men WomenOmaraye JoshuaNo ratings yet
- Microcytic Hypochromic Anemia: - M QariDocument33 pagesMicrocytic Hypochromic Anemia: - M QarirohitNo ratings yet
- Anemia in ChildrenDocument67 pagesAnemia in ChildrenAgung Maulana ArmstrongNo ratings yet
- An Approach To Anemic PatientDocument79 pagesAn Approach To Anemic PatientHussain AzharNo ratings yet
- Anemia of Diminished ErythropoiesisDocument43 pagesAnemia of Diminished ErythropoiesisJared Khoo Er HauNo ratings yet
- Anemia in ChildrenDocument67 pagesAnemia in ChildrenDenny BimatamaNo ratings yet
- Lecture 5.1 Hemolytic Anemia MMDocument41 pagesLecture 5.1 Hemolytic Anemia MMMoeed AliNo ratings yet
- Week 13: Trace Elements Methods and InstrumentationDocument10 pagesWeek 13: Trace Elements Methods and Instrumentationhersey miayoNo ratings yet
- Iron Def ADocument20 pagesIron Def Aacs.pathNo ratings yet
- Genetic Liver DiseasesDocument2 pagesGenetic Liver DiseasesMohammed NaseemuddinNo ratings yet
- AnemiaDocument63 pagesAnemiaRasoulNo ratings yet
- AnaemiaDocument108 pagesAnaemiaakoeljames8543No ratings yet
- Evaluation of The Anemic PatientDocument44 pagesEvaluation of The Anemic PatientShobana KmNo ratings yet
- 1 Anemia PDDocument40 pages1 Anemia PDአንዋርጀማልNo ratings yet
- Approach To Anaemia: S.NdungeDocument30 pagesApproach To Anaemia: S.NdungececiliaNo ratings yet
- HemochromatosisDocument35 pagesHemochromatosisIrshad Ahamed Ilyas100% (1)
- Haematology-Summary My NotesDocument24 pagesHaematology-Summary My NotesToria053No ratings yet
- Liver Cirrhosis, A Simple Guide To The Condition, Treatment And Related DiseasesFrom EverandLiver Cirrhosis, A Simple Guide To The Condition, Treatment And Related DiseasesNo ratings yet
- MCQ FacebookDocument14 pagesMCQ Facebookkays30002403No ratings yet
- Ishemic Heart Disease-y-II-2011 (Student Version)Document51 pagesIshemic Heart Disease-y-II-2011 (Student Version)kays30002403No ratings yet
- Edward L. Lee, M.D. Professor & Chairman Department of Pathology Howard University College of MedicineDocument44 pagesEdward L. Lee, M.D. Professor & Chairman Department of Pathology Howard University College of Medicinekays30002403No ratings yet
- Anemia 2011 Student Dental FDocument64 pagesAnemia 2011 Student Dental Fkays30002403No ratings yet
- MCQSDocument25 pagesMCQSkays30002403No ratings yet
- Pathology of Eye-I-2013-FinalDocument80 pagesPathology of Eye-I-2013-Finalkays30002403No ratings yet
- Glomerulonephritis: Scott Wenderfer Pediatric Renal Fellow November 11, 2004Document56 pagesGlomerulonephritis: Scott Wenderfer Pediatric Renal Fellow November 11, 2004kays30002403No ratings yet
- Extravasation Mucocele - A Case ReportDocument3 pagesExtravasation Mucocele - A Case ReportSSR-IIJLS JournalNo ratings yet
- 1.2 Drug Devt and ProcessDocument14 pages1.2 Drug Devt and ProcessJohn King AmuraoNo ratings yet
- Chemistry Case 1: Chem PanelDocument3 pagesChemistry Case 1: Chem PanelangelNo ratings yet
- Blood Glucose MonitoringDocument10 pagesBlood Glucose MonitoringSarah Jane MaganteNo ratings yet
- Liver & Its Function.Document2 pagesLiver & Its Function.Niraj NiketNo ratings yet
- Amphotericin B Deoxycholate (Conventional) - Drug Information - UpToDate-6Document4 pagesAmphotericin B Deoxycholate (Conventional) - Drug Information - UpToDate-6Vh TRNo ratings yet
- Carbohydrate Nutrition and Team Sport PerformanceDocument10 pagesCarbohydrate Nutrition and Team Sport PerformanceKlaraNo ratings yet
- Monomer Polymer WsDocument2 pagesMonomer Polymer WsclaudNo ratings yet
- The Healy Programs Full List-0001Document15 pagesThe Healy Programs Full List-0001Vikram SharmaNo ratings yet
- 72 Endo AnnotatedDocument9 pages72 Endo AnnotatedErika ArceoNo ratings yet
- Chapter 30 - Endocrine Pancreas and Pharmacotherapy of Diabetes Mellitus and HypoglycemiaDocument12 pagesChapter 30 - Endocrine Pancreas and Pharmacotherapy of Diabetes Mellitus and HypoglycemiaPhúc NguyễnNo ratings yet
- Mohammed H. Eid - The Intensivist-Middle East & ElMarwa For Publishing & Distribution, GN (2019)Document372 pagesMohammed H. Eid - The Intensivist-Middle East & ElMarwa For Publishing & Distribution, GN (2019)Mohamed Moussa100% (1)
- Drug Clearance: Dr. Rajib Bhattacharjee Asstt. Professor Dept of Pharmacy, NSUDocument11 pagesDrug Clearance: Dr. Rajib Bhattacharjee Asstt. Professor Dept of Pharmacy, NSUMohannad AlfadhalNo ratings yet
- Liu2018 NAD Recommended by AttiaDocument20 pagesLiu2018 NAD Recommended by AttiaShantanu JhaNo ratings yet
- Anovulatory InfertilityDocument57 pagesAnovulatory InfertilityUsha AnengaNo ratings yet
- Fabia Ms TellmeDocument2 pagesFabia Ms TellmeDavid Chege K.No ratings yet
- Grade 10 Quarter 3 Lym BDocument4 pagesGrade 10 Quarter 3 Lym BLymberth Benalla100% (1)
- EBCR - Cut Neubi GethaDocument19 pagesEBCR - Cut Neubi GethaNagib MuhammadNo ratings yet
- Renal Disease of Small AnimalsDocument46 pagesRenal Disease of Small AnimalsTahir KasimNo ratings yet
- Adrenergic AntagonistsDocument18 pagesAdrenergic AntagonistsKarina MadriagaNo ratings yet
- (Download PDF) Endocrine Emergencies Recognition and Treatment Lynn Loriaux and Chaim Vanek Online Ebook All Chapter PDFDocument42 pages(Download PDF) Endocrine Emergencies Recognition and Treatment Lynn Loriaux and Chaim Vanek Online Ebook All Chapter PDFsherry.lanzo797100% (14)
- DM Type 2Document13 pagesDM Type 2Yssah Moira HamacNo ratings yet
- Caveats: Acid Base Disorder Paco2 PH Primary Change Compensatory Change EtiologyDocument6 pagesCaveats: Acid Base Disorder Paco2 PH Primary Change Compensatory Change EtiologyPau SorianoNo ratings yet
- Las 1Document5 pagesLas 1rosemarie lingonNo ratings yet
- Biology 22Document15 pagesBiology 22mabdullah12579No ratings yet
- Topic 8 QuestionsDocument90 pagesTopic 8 QuestionsRaiannur RohmanNo ratings yet