Download as pptx, pdf, or txt
You might also like
- Protocol For The Conduct of Stability StudiesDocument4 pagesProtocol For The Conduct of Stability StudiesManish shankarpure100% (1)
- Documentation in Technology TransferDocument8 pagesDocumentation in Technology TransferManish shankarpure100% (1)
- Module 6 - IV Calculations, Solutions, EquipmentDocument3 pagesModule 6 - IV Calculations, Solutions, Equipmentlool89175% (4)
- C - 4 Dosage Form DesignDocument67 pagesC - 4 Dosage Form DesignFelicity TineNo ratings yet
- Stability Study Guidance ProtocolDocument10 pagesStability Study Guidance ProtocolManish shankarpure100% (2)
- SOP For Hot Air OvenDocument1 pageSOP For Hot Air OvenManish shankarpure100% (1)
- Clinical PharmacyDocument38 pagesClinical PharmacyBalakrishna Thalamanchi100% (2)
- Drug and Therapeutics Committee - ToRDocument3 pagesDrug and Therapeutics Committee - ToRFelix Nathan TrisnadiNo ratings yet
- PedoDocument2 pagesPedoHenyo AkoNo ratings yet
- Transdermal Drug Delivery System An OverviewDocument10 pagesTransdermal Drug Delivery System An OverviewJoko RinantoNo ratings yet
- Dosage - Chapter 7Document6 pagesDosage - Chapter 7Kim ManlangitNo ratings yet
- 1 Drug Polymorphism and Dosage Form Design A Practical PerspectiveDocument13 pages1 Drug Polymorphism and Dosage Form Design A Practical Perspectivejulieth vNo ratings yet
- Session 1 Introduction To PICS GMP 009-14Document12 pagesSession 1 Introduction To PICS GMP 009-14Elton SubijanoNo ratings yet
- Introduction To BiopharmaceuticsDocument27 pagesIntroduction To BiopharmaceuticsAmina Akther Mim 1821179649No ratings yet
- Liquid DosageDocument29 pagesLiquid DosageDDS (Dingdong Dantes Supporter)No ratings yet
- Bioavailability: Drug Product PerformanceDocument38 pagesBioavailability: Drug Product PerformancesaadNo ratings yet
- Dosage - Chapter 7Document6 pagesDosage - Chapter 7kaukau4everNo ratings yet
- LiberationDocument33 pagesLiberationAnthony Catapang PascualNo ratings yet
- Solid Dosage FormsDocument4 pagesSolid Dosage Formscofodike1No ratings yet
- Oecd 402Document7 pagesOecd 402Sandro SotomayorNo ratings yet
- Pharmaceutics IntroductionDocument9 pagesPharmaceutics IntroductionVIJAY KUMAR TIRUKKACHINo ratings yet
- Industrial Pharmacy PDFDocument8 pagesIndustrial Pharmacy PDFdrugdrugNo ratings yet
- ICH Guideline Q11 On Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/biological Entities)Document27 pagesICH Guideline Q11 On Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/biological Entities)rodcam1No ratings yet
- C 14 SuspensionDocument119 pagesC 14 SuspensionVinod ChoudharyNo ratings yet
- 634581643032102500Document130 pages634581643032102500DrVenu Madhav KNo ratings yet
- Dosage Form DesignDocument104 pagesDosage Form Designprinceamit100% (1)
- Handbk of Basic Pharmacokinetics Chap. 2Document3 pagesHandbk of Basic Pharmacokinetics Chap. 2MoonGalaxyNo ratings yet
- Profesi Unjani Dispensing TechniquesDocument26 pagesProfesi Unjani Dispensing TechniquesNur AjiNo ratings yet
- Factors Influencing Progressive Utilization of Palliative Care Services Among Cancer Patients in Kenya The Case of Nairobi HospiceDocument50 pagesFactors Influencing Progressive Utilization of Palliative Care Services Among Cancer Patients in Kenya The Case of Nairobi HospiceProficient WritersNo ratings yet
- Bioavailability, Bioequivalence and BCS System: by Dr. Ashwani Kumar VermaDocument35 pagesBioavailability, Bioequivalence and BCS System: by Dr. Ashwani Kumar Vermagopal jhaNo ratings yet
- The Changing Roles of Pharmacists in SocietyDocument13 pagesThe Changing Roles of Pharmacists in SocietyIman AmrNo ratings yet
- Drug Elimination: Termination of Drug ActionDocument80 pagesDrug Elimination: Termination of Drug ActionLidyaNo ratings yet
- 6 SyrupsDocument37 pages6 Syrups鄭宇揚No ratings yet
- Lipinski Rule of FiveDocument21 pagesLipinski Rule of FiveSasikala RajendranNo ratings yet
- Design of Dosage FormsDocument17 pagesDesign of Dosage FormsMuhammad HilmiNo ratings yet
- Intro Drug DiscoveryDocument32 pagesIntro Drug Discoveryapi-19965961No ratings yet
- Diff Osmosis Lab Sp11Document8 pagesDiff Osmosis Lab Sp11Kelly TrainorNo ratings yet
- Cosmetic Lecture-2Document7 pagesCosmetic Lecture-2Daddy RulesNo ratings yet
- Uniformity Dosage Unit USPDocument4 pagesUniformity Dosage Unit USPwike marelitaNo ratings yet
- ADRDocument34 pagesADRShaikh FaseehuddinNo ratings yet
- Class 17-2-3 Factorial DesignDocument26 pagesClass 17-2-3 Factorial Designb_shadid8399No ratings yet
- Pre FormulationDocument13 pagesPre FormulationCm MouliNo ratings yet
- Excipients ProfileDocument7 pagesExcipients ProfileTamilarasan DeenathayalanNo ratings yet
- Pharmaceutical MeasurementDocument26 pagesPharmaceutical MeasurementChengDNo ratings yet
- Prescription IncompatibilitiesDocument22 pagesPrescription IncompatibilitiesSunshine BaclaanNo ratings yet
- QC Tests For Tablet Dosage FormsDocument8 pagesQC Tests For Tablet Dosage FormsSai Krishna ManchikantiNo ratings yet
- 4-Drug Delivery Systems (Autosaved)Document41 pages4-Drug Delivery Systems (Autosaved)Chelle PaloNo ratings yet
- Lecture - 1 Dosage FormDocument13 pagesLecture - 1 Dosage FormAshique Farhad100% (1)
- Chapter 7. Pharmacokinetics of Oral AbsorptionDocument28 pagesChapter 7. Pharmacokinetics of Oral AbsorptionbencleeseNo ratings yet
- Dosage - Chapter 9Document5 pagesDosage - Chapter 9Kim Manlangit100% (1)
- Factors Influencing Drug Absorption Though Git PDFDocument59 pagesFactors Influencing Drug Absorption Though Git PDFRamakant JoshiNo ratings yet
- IpqcDocument37 pagesIpqcAjitha AzhakesanNo ratings yet
- Pharmacology in Drug DiscoveryDocument26 pagesPharmacology in Drug DiscoveryManuel Christopher MontesclarosNo ratings yet
- DistributionDocument45 pagesDistributionKailas Mali0% (1)
- Pharmaceutical Development and Compatibility Studies On Cytarabine InjectionDocument4 pagesPharmaceutical Development and Compatibility Studies On Cytarabine InjectionAmit KhuntNo ratings yet
- Group 5 Pdis211 Lab E1Document22 pagesGroup 5 Pdis211 Lab E1Mikaela Kean JoseNo ratings yet
- Incompatibility IV AdmixtureDocument17 pagesIncompatibility IV AdmixtureClara Herlina100% (1)
- Pharmacoinformatics: A Tool For Drug DiscoveryDocument29 pagesPharmacoinformatics: A Tool For Drug Discoveryahirebhupendra100% (1)
- Ethical Issues in International Health ResearchDocument36 pagesEthical Issues in International Health ResearchMaicel MonzonNo ratings yet
- 2 USP - OSD Quality TestsDocument6 pages2 USP - OSD Quality TestsSpectre SpectreNo ratings yet
- Dosage FormDocument6 pagesDosage FormAmaila Ch100% (1)
- International Conference On HarmonizationDocument35 pagesInternational Conference On HarmonizationFarah Aman Khan100% (2)
- Quality GuidelinesDocument4 pagesQuality GuidelinesSrinivasNo ratings yet
- Quality GuidelinesDocument4 pagesQuality GuidelinesSrinivasNo ratings yet
- ICH ListDocument7 pagesICH ListROHIT CONSULTANCYNo ratings yet
- Ich Guidelines: #Fouziya's-NotesDocument3 pagesIch Guidelines: #Fouziya's-NotesFouziya BegumNo ratings yet
- QBD Putting Theory Into Practice - Chp2 THE REGULATORY FRAMEWORK PDFDocument26 pagesQBD Putting Theory Into Practice - Chp2 THE REGULATORY FRAMEWORK PDFhdmnauNo ratings yet
- Manish Acts and Rules MindmapDocument2 pagesManish Acts and Rules MindmapManish shankarpureNo ratings yet
- Swami Ramanand Teerth Marathwada University, Nanded: Faculty of Pharmaceutical Sciences and TechnologyDocument23 pagesSwami Ramanand Teerth Marathwada University, Nanded: Faculty of Pharmaceutical Sciences and TechnologyManish shankarpureNo ratings yet
- Regulatory Affairs SagarDocument1 pageRegulatory Affairs SagarManish shankarpureNo ratings yet
- SOP For Hot Air OvenDocument1 pageSOP For Hot Air OvenManish shankarpureNo ratings yet
- Uv - Visible SpectrosDocument21 pagesUv - Visible SpectrosManish shankarpure100% (1)
- IPCA LabsDocument25 pagesIPCA Labsneekuj malikNo ratings yet
- Front PagesDocument7 pagesFront PagesAditya JainNo ratings yet
- India The Emerging Hub For Biologics and BiosimilarsDocument22 pagesIndia The Emerging Hub For Biologics and BiosimilarssanjitlNo ratings yet
- Daftar Nama Obat SIMPUSDocument11 pagesDaftar Nama Obat SIMPUSsdk.achmad TubeNo ratings yet
- B Pharmacy 2015Document114 pagesB Pharmacy 2015GalataNo ratings yet
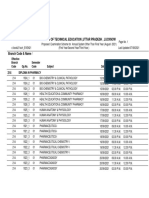
- Board of Technical Education, Uttar Pradesh, Lucknow: Branch Code QP - No. Semester Code Subject Date Timings EffectiveDocument2 pagesBoard of Technical Education, Uttar Pradesh, Lucknow: Branch Code QP - No. Semester Code Subject Date Timings Effectivealice HNo ratings yet
- Drug StudyDocument7 pagesDrug StudyHanidah RahimNo ratings yet
- Model Pharmacy in BangladeshDocument7 pagesModel Pharmacy in BangladeshMoinuddin Khan KafiNo ratings yet
- Hetero Corp Presentation 2016Document23 pagesHetero Corp Presentation 2016Evan TanswariNo ratings yet
- Ebook Management of Safety Information From Clinical Trials Report of CIOMS Working Group VIDocument306 pagesEbook Management of Safety Information From Clinical Trials Report of CIOMS Working Group VINalendra NalendraNo ratings yet
- Chapter 5 - Documentation of Patient Encounters and InterventionsDocument16 pagesChapter 5 - Documentation of Patient Encounters and InterventionsV MNo ratings yet
- Chapter 11 Multiple Dosage RegimenDocument35 pagesChapter 11 Multiple Dosage RegimenYuli Irvaransiah DIatun NIkmah100% (2)
- V30N9 PDFDocument6 pagesV30N9 PDFMargarito jrNo ratings yet
- Injeksi & Oplosan ObatDocument16 pagesInjeksi & Oplosan ObatFebriani RatnaNo ratings yet
- Pgdwash021 - Sparsh Hospital - Sanitation and Public HealthDocument9 pagesPgdwash021 - Sparsh Hospital - Sanitation and Public HealthMadhumithaa muralieNo ratings yet
- Risperidone Risperdal Oral Injection Equivalent GuidanceDocument3 pagesRisperidone Risperdal Oral Injection Equivalent GuidancemaladominiNo ratings yet
- Del Rosario v. BengzonDocument7 pagesDel Rosario v. BengzonLee YuNo ratings yet
- Cytostatics As HazardousDocument19 pagesCytostatics As HazardousIvan PerezNo ratings yet
- SRM Valliammai Engineering College (An Autonomous Institution)Document11 pagesSRM Valliammai Engineering College (An Autonomous Institution)Renit AntoNo ratings yet
- Content Owner: Chetan Kothari Compiled By: Abhishek MurarkaDocument25 pagesContent Owner: Chetan Kothari Compiled By: Abhishek MurarkaJ.BNo ratings yet
- PSUK Dispensary SOPS ToolkitDocument10 pagesPSUK Dispensary SOPS ToolkitHasif BastiarNo ratings yet
- Journal Reflection in Immersion Pharmacy LaboratoryDocument4 pagesJournal Reflection in Immersion Pharmacy LaboratoryCiel GevenNo ratings yet
- Community Pharmacy Layout Full ChapterDocument4 pagesCommunity Pharmacy Layout Full ChaptersaleemNo ratings yet
- Medication GuidesDocument38 pagesMedication GuidesEko YuliantoNo ratings yet
- Teori-Teori Perubahan PerilakuDocument32 pagesTeori-Teori Perubahan PerilakuJulham Arya S KedNo ratings yet
- Minutes of 290th Meeting of Registration BoardDocument1,286 pagesMinutes of 290th Meeting of Registration BoardUsman DarNo ratings yet