Download as ppt, pdf, or txt
You might also like
- Chapter 1 Glass IndustryDocument32 pagesChapter 1 Glass Industryد.حاتممرقه100% (1)
- Characteristic of HybridizationDocument8 pagesCharacteristic of HybridizationMehul KapoorNo ratings yet
- Komlexemaster 7 SemDocument49 pagesKomlexemaster 7 SemRaheem SimsNo ratings yet
- Lectures 3 4, Nomenclature of Coordination Complexes PDFDocument6 pagesLectures 3 4, Nomenclature of Coordination Complexes PDFFahed DawodNo ratings yet
- Transition Metals and Coordination ComplexesDocument61 pagesTransition Metals and Coordination ComplexesGliezl ImperialNo ratings yet
- 152 C 20 NewDocument37 pages152 C 20 NewGaotingweNo ratings yet
- Tatanama Senyawa KompleksDocument52 pagesTatanama Senyawa KompleksRomanNo ratings yet
- Transition Metals and Coordination ChemistryDocument77 pagesTransition Metals and Coordination ChemistryAdistaNo ratings yet
- Transition Metals and Coordination ChemistryDocument37 pagesTransition Metals and Coordination ChemistryMortey Yunus DoeNo ratings yet
- Haloareans and Halo AlkanesDocument23 pagesHaloareans and Halo Alkanesthinkiit100% (1)
- Nomenclature of Coordination Complexes PDFDocument10 pagesNomenclature of Coordination Complexes PDFHamxa KhanNo ratings yet
- Rule 1:: Na (PTCL (NH) ) K (Cubr)Document10 pagesRule 1:: Na (PTCL (NH) ) K (Cubr)Hamxa KhanNo ratings yet
- Study Unit 3 - 3.1 - Naming-1-1Document19 pagesStudy Unit 3 - 3.1 - Naming-1-1ABDUL MOHAMEDNo ratings yet
- Revision Notes On Co-Ordination CompoundsDocument12 pagesRevision Notes On Co-Ordination CompoundsAnonymous vRpzQ2BLNo ratings yet
- Crystal Field Teory Coordination ChemistryDocument103 pagesCrystal Field Teory Coordination ChemistryM Faqri Fahrozi HNo ratings yet
- Inorganic Chapter19Document23 pagesInorganic Chapter19barkatullah0% (1)
- Transition Metal ComplexesDocument103 pagesTransition Metal Complexesidownloadbooksforstu100% (1)
- Lecture 3Document11 pagesLecture 3saboor_91No ratings yet
- Coordination ChemistryDocument14 pagesCoordination Chemistryjangidudit115No ratings yet
- Chem261 Part 1-A and BDocument33 pagesChem261 Part 1-A and BAriana NaidooNo ratings yet
- D-Block Metal Chemistry: General ConsiderationsDocument23 pagesD-Block Metal Chemistry: General ConsiderationsPrativa BeheraNo ratings yet
- 4-Coordination Chemistry IDocument61 pages4-Coordination Chemistry Igunjan pratapNo ratings yet
- Paper 12 3983 24Document6 pagesPaper 12 3983 24bsmalah11alroxnamNo ratings yet
- 1CHEM261 Part1A WVZDocument30 pages1CHEM261 Part1A WVZndlovumpendulo281No ratings yet
- Co-Ordination CompoundsDocument11 pagesCo-Ordination CompoundsShashank AgarwalNo ratings yet
- Coordination Compounds 1Document12 pagesCoordination Compounds 1Anonymous 8VJhV1eI2y100% (3)
- CH 9Document13 pagesCH 9Tr Mazhar PunjabiNo ratings yet
- Book 0chapter 15Document20 pagesBook 0chapter 15Bich Hue NguyenNo ratings yet
- Coordination CompoundDocument6 pagesCoordination CompoundniyojetNo ratings yet
- C I & C C: Omplex ONS Oordination OmpoundsDocument2 pagesC I & C C: Omplex ONS Oordination OmpoundsAL-AMEENNo ratings yet
- Nomenclature of 6 - 2018 - 11 - 04!09 - 13 - 23 - PMDocument9 pagesNomenclature of 6 - 2018 - 11 - 04!09 - 13 - 23 - PMnoor ul ainNo ratings yet
- Theory EnglishDocument11 pagesTheory Englishd anjilappaNo ratings yet
- Chem Chap 5 Coordination CompoundsDocument71 pagesChem Chap 5 Coordination Compoundsissacpaul382No ratings yet
- SCH 1201 - Inorganic Chemistry Ii - Transition Metal ChemistryDocument305 pagesSCH 1201 - Inorganic Chemistry Ii - Transition Metal ChemistrysanelisofuturemoyoNo ratings yet
- Coordination Compounds 2Document48 pagesCoordination Compounds 2pavithra KumarNo ratings yet
- Nomenclature of CoordinationcompoundsDocument7 pagesNomenclature of CoordinationcompoundsPravin NegiNo ratings yet
- Coordination Chemistry NamingDocument22 pagesCoordination Chemistry NamingPadirikuppam PavithraNo ratings yet
- Coordination Chemistry I Structures Isomers and Naming ComplexesDocument110 pagesCoordination Chemistry I Structures Isomers and Naming ComplexesshengNo ratings yet
- Coordination Copounds NotesDocument22 pagesCoordination Copounds NotesGaurav SinghaNo ratings yet
- Coordination Compounds IDocument58 pagesCoordination Compounds IAli Muhammad Kamba60% (5)
- Transition Elements PDFDocument18 pagesTransition Elements PDFArslanAliNo ratings yet
- Chap 9part1Document69 pagesChap 9part1Marie Kris NogaNo ratings yet
- UNIT 9 Topic: Coordination CompoundsDocument9 pagesUNIT 9 Topic: Coordination CompoundsDeva RajNo ratings yet
- Coordination CompoundsDocument51 pagesCoordination CompoundsasdfNo ratings yet
- Chapter 9 Co Ordination CompoundsDocument46 pagesChapter 9 Co Ordination Compoundssukaina fatimaNo ratings yet
- Comple JosDocument25 pagesComple JosMarco Antonio FloresNo ratings yet
- Activity 22: Monodentate One With Two Such Atoms Is Bidentate, One With Three Such Atoms Is Tridentate, EtcDocument8 pagesActivity 22: Monodentate One With Two Such Atoms Is Bidentate, One With Three Such Atoms Is Tridentate, EtcAhmed MahmoudNo ratings yet
- 12 Chemistry Impq CH07 The P Block Elements 03Document15 pages12 Chemistry Impq CH07 The P Block Elements 03Ricky SharmaNo ratings yet
- Coordination CompdsDocument7 pagesCoordination CompdsAnil BahriNo ratings yet
- Inorganic Paper III 2010Document47 pagesInorganic Paper III 2010ls1955100% (3)
- Hsslive Xii Chemistry CH 9 Coordination Compounds by SajeevDocument2 pagesHsslive Xii Chemistry CH 9 Coordination Compounds by SajeevrasalgafoorrvgNo ratings yet
- Chem NotesDocument17 pagesChem NotesCND PurPNo ratings yet
- R K Sharma Coordination Chemistry 10-30Document23 pagesR K Sharma Coordination Chemistry 10-30Kamal KishoreNo ratings yet
- Topic 7 - Coordination ChemistryDocument30 pagesTopic 7 - Coordination ChemistryRex JusayanNo ratings yet
- Coordination Chemistry: Hui LiDocument91 pagesCoordination Chemistry: Hui LiPdssnNo ratings yet
- Coordination CompoundDocument19 pagesCoordination CompounddeepjoypalNo ratings yet
- Naming Coordination CompoundsDocument8 pagesNaming Coordination CompoundsBLi'H'AbieeNo ratings yet
- Class 12 Chapter 9 Coordination CompoundsDocument66 pagesClass 12 Chapter 9 Coordination Compoundsmishraaryan954No ratings yet
- Nomenclature of Coordination ComplexesDocument6 pagesNomenclature of Coordination ComplexesChemo_Eldaly_4662No ratings yet
- Chemistry: a QuickStudy Laminated Reference GuideFrom EverandChemistry: a QuickStudy Laminated Reference GuideRating: 4.5 out of 5 stars4.5/5 (2)
- Why Are Chemicals Not Named John? Naming Chemical Compounds 6th Grade | Children's Chemistry BooksFrom EverandWhy Are Chemicals Not Named John? Naming Chemical Compounds 6th Grade | Children's Chemistry BooksNo ratings yet
- Vibrational Spectra of Organometallics: Theoretical and Experimental DataFrom EverandVibrational Spectra of Organometallics: Theoretical and Experimental DataNo ratings yet
- Hebron University College of Science and Technology Department of ChemistryDocument3 pagesHebron University College of Science and Technology Department of Chemistryد.حاتممرقهNo ratings yet
- Compr Solid State Chemistry Questions and AnswersDocument44 pagesCompr Solid State Chemistry Questions and Answersد.حاتممرقهNo ratings yet
- Materials Science Questions and AnswersDocument7 pagesMaterials Science Questions and Answersد.حاتممرقهNo ratings yet
- MM 302 X Ray Mcqs Test Combined - 1 4Document16 pagesMM 302 X Ray Mcqs Test Combined - 1 4د.حاتممرقه100% (6)
- Solid State Chemistry Questions and AnswersDocument14 pagesSolid State Chemistry Questions and Answersد.حاتممرقهNo ratings yet
- Ceramic IndustriesDocument9 pagesCeramic Industriesد.حاتممرقهNo ratings yet
- Hebron University College of Science and Technology Department of ChemistryDocument4 pagesHebron University College of Science and Technology Department of Chemistryد.حاتممرقهNo ratings yet
- Cement Industry: Manufacturing Process of Portland Cement Raw MaterialsDocument11 pagesCement Industry: Manufacturing Process of Portland Cement Raw Materialsد.حاتممرقهNo ratings yet
- P Blockelements 1608Document51 pagesP Blockelements 1608د.حاتممرقهNo ratings yet
- Ceramic Water Purifier: Performance Data SheetDocument7 pagesCeramic Water Purifier: Performance Data SheetT.BieniekNo ratings yet
- Lesson 11 Dative Covalent (Or Coordinate Bonding)Document22 pagesLesson 11 Dative Covalent (Or Coordinate Bonding)dela2No ratings yet
- Manganese Geolite PDFDocument2 pagesManganese Geolite PDFShah Newaz KabirNo ratings yet
- R Process Nucleosynthesis and Its Site: Mario A. Riquelme Theore Cal Seminar Fall 2009Document22 pagesR Process Nucleosynthesis and Its Site: Mario A. Riquelme Theore Cal Seminar Fall 2009M Yudi SuhendarNo ratings yet
- English 1000 Word EssayDocument4 pagesEnglish 1000 Word Essayapi-320830647No ratings yet
- Fantastic A (Seventh) List of New Mineral Names 2 Mineralogical SocietDocument19 pagesFantastic A (Seventh) List of New Mineral Names 2 Mineralogical SocietLaura BecerraNo ratings yet
- Grade 9 - Ionic BondingDocument80 pagesGrade 9 - Ionic BondingVictoria Lowman67% (3)
- 9 Tausends GoldDocument14 pages9 Tausends GoldGeorge O AmadoNo ratings yet
- The Chemistry of Alkaly AmidesDocument69 pagesThe Chemistry of Alkaly AmidesGustavoNo ratings yet
- CML 100: Inorganic Chemistry Contents: Bonding in Transition Metal ComplexesDocument19 pagesCML 100: Inorganic Chemistry Contents: Bonding in Transition Metal Complexestushar guptaNo ratings yet
- Identify The Presence of Protein in Egg AlbuminDocument2 pagesIdentify The Presence of Protein in Egg AlbuminManraj Singh RoopraNo ratings yet
- No. Jenis Plate Size Coating Material Coating Thickness One Sides Surface Finish Chemical Treatment Vendor StandardDocument3 pagesNo. Jenis Plate Size Coating Material Coating Thickness One Sides Surface Finish Chemical Treatment Vendor StandardDimas A PrakosoNo ratings yet
- Organic Compound: Definitions of Organic Vs Inorganic HistoryDocument5 pagesOrganic Compound: Definitions of Organic Vs Inorganic HistoryAdvait BurandeNo ratings yet
- Acids & Bases Strength PredictionDocument15 pagesAcids & Bases Strength Predictionjudyli990% (1)
- Thermals Pray Material GuideDocument132 pagesThermals Pray Material GuideKyeong Cheol LeeNo ratings yet
- Electrochemistry MCQ SendDocument7 pagesElectrochemistry MCQ SendRajendra ChikkamathNo ratings yet
- Complexometric Titration PDFDocument11 pagesComplexometric Titration PDFDr. Gunjan SarkarNo ratings yet
- Note On Atoms, Molecules, Valency and RadicalsDocument3 pagesNote On Atoms, Molecules, Valency and RadicalsRadiant BrothersNo ratings yet
- Sku3073 Experiment 3Document8 pagesSku3073 Experiment 3MUHAMMAD ARIF AIMAN BIN MOKHTARNo ratings yet
- Atoms, Ions, MoleculesDocument11 pagesAtoms, Ions, MoleculesGracia Thalia TNo ratings yet
- KS4 Noble GasesDocument15 pagesKS4 Noble Gasesaderifaldi88No ratings yet
- SPM Practice Chap3 F4Document7 pagesSPM Practice Chap3 F4Shervin Fernandez0% (1)
- EDTA 2NA Analysis MethodDocument6 pagesEDTA 2NA Analysis MethodClayton UkracheskiNo ratings yet
- INJSO Answer Key & SolutionDocument5 pagesINJSO Answer Key & SolutionYatish Goyal100% (1)
- 1 BondingDocument50 pages1 BondingSherey FathimathNo ratings yet
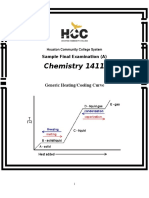
- Chemistry 1411: Generic Heating/Cooling CurveDocument19 pagesChemistry 1411: Generic Heating/Cooling CurveKinal PatelNo ratings yet
- Discharge Standards For DubaiDocument6 pagesDischarge Standards For DubaiHRK65No ratings yet
- Su 1975Document2 pagesSu 1975thoriqalf12No ratings yet
- Mat ListDocument35 pagesMat ListhaithamNo ratings yet