
Physical Properties of Proteins
Physical Properties of Proteins
You might also like
- Ultimate Guide To The CSCS ExamDocument104 pagesUltimate Guide To The CSCS Examrobertobarron88% (17)
- Genetic Bypass BookDocument319 pagesGenetic Bypass BookAngie Merceir100% (6)
- Built To Endure3Document208 pagesBuilt To Endure3nichoff100% (2)
- Protein Metabolism and Acids SamyA1Document170 pagesProtein Metabolism and Acids SamyA1Abby RahmanNo ratings yet
- Effects of Creatine Supplementation On Renal Function - A Systematic Review and Meta-AnalysisDocument10 pagesEffects of Creatine Supplementation On Renal Function - A Systematic Review and Meta-AnalysisNaiara CaramuruNo ratings yet
- Protien and Urea CycleDocument33 pagesProtien and Urea CycleTAUQEER Ali shahNo ratings yet
- Urea Cycle LehningerDocument34 pagesUrea Cycle LehningerMohamadJamaludinNo ratings yet
- Urea Cycle and Its DefectsDocument48 pagesUrea Cycle and Its DefectsStevia NdoeNo ratings yet
- Protein Metabolism Dental and Physiotherapy Part 1Document17 pagesProtein Metabolism Dental and Physiotherapy Part 1Nada Atef KoraitemNo ratings yet
- Urea Cycle and Protein MetabolismDocument39 pagesUrea Cycle and Protein Metabolismikramullahkhan211No ratings yet
- Urea Cycle & HyperammoniaDocument6 pagesUrea Cycle & HyperammoniaSal TlsNo ratings yet
- 1.15 Amino Acid MetabolismDocument16 pages1.15 Amino Acid MetabolismnkhomaslaterNo ratings yet
- Catabolism of Proteins and Amino AcidsDocument63 pagesCatabolism of Proteins and Amino Acidsflairtique shopNo ratings yet
- UrineDocument41 pagesUrineWahab KhaniNo ratings yet
- Chapter X - Mechanism of Protein MetabolismDocument30 pagesChapter X - Mechanism of Protein MetabolismAngelo AngelesNo ratings yet
- Amino Acid MetabolismDocument23 pagesAmino Acid MetabolismZ ZNo ratings yet
- Metabolism of Aminoacids 2Document68 pagesMetabolism of Aminoacids 2Mi PatelNo ratings yet
- Amino Acids Metabolism (Catabolism) and Clinical SignificantDocument33 pagesAmino Acids Metabolism (Catabolism) and Clinical SignificantBehailu TsegayeNo ratings yet
- Bio - Urea CycleDocument57 pagesBio - Urea CycleMahmoud hilmyNo ratings yet
- Protein MetaboilisamDocument18 pagesProtein MetaboilisamSumit PandyaNo ratings yet
- Amino Acid Metabolism II. Urea CycleDocument39 pagesAmino Acid Metabolism II. Urea Cycleputri jessicaNo ratings yet
- Amino Acid Catabolism-Part-1: Biochemistry For Medics - Lecture Notes Professor (DR.) Namrata ChhabraDocument43 pagesAmino Acid Catabolism-Part-1: Biochemistry For Medics - Lecture Notes Professor (DR.) Namrata Chhabrashree devNo ratings yet
- Urea Cycle and Disorders - RM - F2014Document18 pagesUrea Cycle and Disorders - RM - F2014Leon WarrenNo ratings yet
- Individual Amino Acid 2Document52 pagesIndividual Amino Acid 2Mai Elsayed OmaraNo ratings yet
- UreaDocument16 pagesUreaMuhammad AmjadNo ratings yet
- Protein Turn OverDocument29 pagesProtein Turn Overabdullah zaheerNo ratings yet
- AA Metabolism HANDOUTDocument4 pagesAA Metabolism HANDOUTnajwa hajjaliNo ratings yet
- Amino Acid CatabolismDocument19 pagesAmino Acid Catabolismwmdpr4x64fNo ratings yet
- Protein and Amino Acid MetabolismDocument52 pagesProtein and Amino Acid MetabolismRisky OpponentNo ratings yet
- Katabolisme Protein Dan Asam AmimoDocument35 pagesKatabolisme Protein Dan Asam AmimoLha YhoeLaNo ratings yet
- Urea CycleDocument39 pagesUrea Cycledrismailkm20No ratings yet
- Aminoacid MetabolismDocument22 pagesAminoacid MetabolismFazal Akbar KhaliliNo ratings yet
- Item - 11, Protein MetabolismDocument12 pagesItem - 11, Protein MetabolismSheikh FahadNo ratings yet
- 3 Metabolism of Proteins & Amino AcidsDocument79 pages3 Metabolism of Proteins & Amino AcidsYashfa YasinNo ratings yet
- Urea CycleDocument4 pagesUrea Cycletfgrn7srtqNo ratings yet
- Introduction Amino Acid Matabolism and CatabolismDocument45 pagesIntroduction Amino Acid Matabolism and CatabolismAboubakar Moalim Mahad moh'd100% (1)
- Protein MetabolismDocument182 pagesProtein MetabolismSimra ZahidNo ratings yet
- Amino Acid MetabolismDocument35 pagesAmino Acid MetabolismRajkishor YadavNo ratings yet
- Proteins and Amino Acids Metabolism: Tahun Ajar 2016/2017Document47 pagesProteins and Amino Acids Metabolism: Tahun Ajar 2016/2017Aswar AyuNo ratings yet
- Biochemistry Lecture PPT 7Document16 pagesBiochemistry Lecture PPT 7Chiranjeevi JoshiNo ratings yet
- Amino Acid & Protein MetabolismDocument59 pagesAmino Acid & Protein MetabolismAndualemNo ratings yet
- Other Amino Acids Form Succinyl CoaDocument23 pagesOther Amino Acids Form Succinyl CoanyarieNo ratings yet
- Ch18 - Amino Acid Oxidation Production of UreaDocument50 pagesCh18 - Amino Acid Oxidation Production of UreaSalma KhoirunnisaNo ratings yet
- BIO 202 Biochemistry II by Seyhun YURDUGÜL: Amino Acid Metabolism I: Amino Acid BiosynthesisDocument65 pagesBIO 202 Biochemistry II by Seyhun YURDUGÜL: Amino Acid Metabolism I: Amino Acid BiosynthesisYasin Çağrı KılıçerNo ratings yet
- Lecture 5 Protein Metabolism 1Document29 pagesLecture 5 Protein Metabolism 1Elizabeth LagunasNo ratings yet
- BP U9d Protein MetabolismDocument80 pagesBP U9d Protein MetabolismChristian Angelo AgbunagNo ratings yet
- Protein and Amino Acid MetabolismDocument32 pagesProtein and Amino Acid MetabolismVirag0% (1)
- Urea CycleDocument4 pagesUrea CycleKunal DuttaNo ratings yet
- 15BT103 Biochem-UNIT 3Document53 pages15BT103 Biochem-UNIT 3Adityanair RA1711009010128No ratings yet
- Urea CycleDocument3 pagesUrea CycleSundaralingam RajNo ratings yet
- General Protein Metabolism NUB Full 2022Document11 pagesGeneral Protein Metabolism NUB Full 2022PIH SHTNo ratings yet
- Biochemistry Week 8 Amino Acid MetabolismDocument29 pagesBiochemistry Week 8 Amino Acid MetabolismKaren 3No ratings yet
- Amino Acid MetabolismDocument81 pagesAmino Acid Metabolismcharuss.394No ratings yet
- Biochemistry: Dr. Dra. Trini Suryowati, MsDocument49 pagesBiochemistry: Dr. Dra. Trini Suryowati, MsDaud ParluhutanNo ratings yet
- Urea Cycle: Dr. Amro Yousef Al-AmlehDocument45 pagesUrea Cycle: Dr. Amro Yousef Al-AmlehDr. Amro YousefNo ratings yet
- Urea Cycle 2Document21 pagesUrea Cycle 2Akinrotimi OluwadunsinNo ratings yet
- Urea CycleDocument8 pagesUrea CycleManohar PattarNo ratings yet
- (Krebs-Henseleit Cycle) : Urea FormationDocument15 pages(Krebs-Henseleit Cycle) : Urea Formationعلي عبيد العتابيNo ratings yet
- B. Katabolisme Asam Amino-1Document19 pagesB. Katabolisme Asam Amino-1M Sifal MaulanaNo ratings yet
- PROTEIN METABOLISM Dea Farha Fira Darson FineDocument44 pagesPROTEIN METABOLISM Dea Farha Fira Darson FineFarhati Mardhiyah100% (1)
- Explain The Ff. Manifestations in The Different Cases That Are Discused BelowDocument29 pagesExplain The Ff. Manifestations in The Different Cases That Are Discused BelowRodel Paulo Tangunan GarciaNo ratings yet
- Amino Acids Metabolism Final For Pharm 2014Document57 pagesAmino Acids Metabolism Final For Pharm 2014Getu LuchesaNo ratings yet
- Catabolism of Amino Acids: DR - Ula Abbas ZekiDocument49 pagesCatabolism of Amino Acids: DR - Ula Abbas ZekinameNo ratings yet
- Protein NeedsDocument9 pagesProtein NeedsPaul BugejaNo ratings yet
- Bio-Inorganic ChemistryDocument66 pagesBio-Inorganic ChemistryPhalynxNo ratings yet
- The Effects of Creatine Ethyl Ester Supplementation Combined With Heavy Resistance Training On Body Composition, Muscle Performance, and Serum and Muscle Creatine LevelsDocument15 pagesThe Effects of Creatine Ethyl Ester Supplementation Combined With Heavy Resistance Training On Body Composition, Muscle Performance, and Serum and Muscle Creatine LevelsMarcio PertileNo ratings yet
- CC Lab 6 TransesDocument6 pagesCC Lab 6 TransesCiara PamonagNo ratings yet
- Non-Protein Nitrogenous Substances: Urea CreatineDocument2 pagesNon-Protein Nitrogenous Substances: Urea CreatineLOU BLESSY CASINONo ratings yet
- Creapure CreatineDocument4 pagesCreapure CreatineAlex RogersNo ratings yet
- How To Inhibit Myostatin For Muscle Growth and Longer LifeDocument5 pagesHow To Inhibit Myostatin For Muscle Growth and Longer Lifedarthlucasx100% (3)
- Revive Stronger Get Big Stay Lean A Comprehensive Guide To Gaining Muscle PDFDocument24 pagesRevive Stronger Get Big Stay Lean A Comprehensive Guide To Gaining Muscle PDFCyrus Dickson A CruzNo ratings yet
- Non-Protein Nitrogenous Constituents of Blood - Urea, Uric Acid EtcDocument50 pagesNon-Protein Nitrogenous Constituents of Blood - Urea, Uric Acid EtcBobskinnyNo ratings yet
- Muscle Science RoundupDocument165 pagesMuscle Science RoundupAlessandro Valentim100% (2)
- Creatine SatbilityDocument2 pagesCreatine SatbilitymonstermuddyNo ratings yet
- Biohacking LiteDocument25 pagesBiohacking Litetxboxi23No ratings yet
- Work Out: To Win BIG!Document153 pagesWork Out: To Win BIG!Alessandro ValentimNo ratings yet
- 2013-UKChO-Round 1 - tcm1Document12 pages2013-UKChO-Round 1 - tcm1vermouth020No ratings yet
- Opt' Eijnde 2001Document6 pagesOpt' Eijnde 2001Prefeitura de FloresNo ratings yet
- Challenge Yourself: Beginner Mens Challenge Preparation GuideDocument23 pagesChallenge Yourself: Beginner Mens Challenge Preparation GuideUni TabberNo ratings yet
- Supplement Education For FireDocument7 pagesSupplement Education For Fireapi-530327871No ratings yet
- Disclaimer: Provided by University of Gloucestershire Research RepositoryDocument34 pagesDisclaimer: Provided by University of Gloucestershire Research Repositorynose que no seNo ratings yet
- Muscular Development August 2015 Usapdf CompressDocument180 pagesMuscular Development August 2015 Usapdf CompressSaurav VermaNo ratings yet
- Nutrition and Hydration For HandballDocument23 pagesNutrition and Hydration For HandballGiorgianaNo ratings yet
- Nutrition Guide by Calisthenics FamilyDocument25 pagesNutrition Guide by Calisthenics FamilyTommy MartelNo ratings yet
- NPNDocument42 pagesNPNreynanrolleNo ratings yet
- Creatine: The Power Supplement?Document13 pagesCreatine: The Power Supplement?api-315070214No ratings yet
- ENERGY ZONES IN SWIMMING. Genadijus Sokolovas, Ph.D. Global Sport Technology, IncDocument6 pagesENERGY ZONES IN SWIMMING. Genadijus Sokolovas, Ph.D. Global Sport Technology, IncMateusNo ratings yet
- PretestDocument3 pagesPretestAlili DudzNo ratings yet
- Nieuw 3Document12 pagesNieuw 3Jasmijn AlmuliNo ratings yet











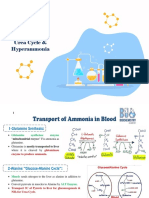












































































Download as pptx, pdf, or txt
You might also like
- Ultimate Guide To The CSCS ExamDocument104 pagesUltimate Guide To The CSCS Examrobertobarron88% (17)
- Genetic Bypass BookDocument319 pagesGenetic Bypass BookAngie Merceir100% (6)
- Built To Endure3Document208 pagesBuilt To Endure3nichoff100% (2)
- Protein Metabolism and Acids SamyA1Document170 pagesProtein Metabolism and Acids SamyA1Abby RahmanNo ratings yet
- Effects of Creatine Supplementation On Renal Function - A Systematic Review and Meta-AnalysisDocument10 pagesEffects of Creatine Supplementation On Renal Function - A Systematic Review and Meta-AnalysisNaiara CaramuruNo ratings yet
- Protien and Urea CycleDocument33 pagesProtien and Urea CycleTAUQEER Ali shahNo ratings yet
- Urea Cycle LehningerDocument34 pagesUrea Cycle LehningerMohamadJamaludinNo ratings yet
- Urea Cycle and Its DefectsDocument48 pagesUrea Cycle and Its DefectsStevia NdoeNo ratings yet
- Protein Metabolism Dental and Physiotherapy Part 1Document17 pagesProtein Metabolism Dental and Physiotherapy Part 1Nada Atef KoraitemNo ratings yet
- Urea Cycle and Protein MetabolismDocument39 pagesUrea Cycle and Protein Metabolismikramullahkhan211No ratings yet
- Urea Cycle & HyperammoniaDocument6 pagesUrea Cycle & HyperammoniaSal TlsNo ratings yet
- 1.15 Amino Acid MetabolismDocument16 pages1.15 Amino Acid MetabolismnkhomaslaterNo ratings yet
- Catabolism of Proteins and Amino AcidsDocument63 pagesCatabolism of Proteins and Amino Acidsflairtique shopNo ratings yet
- UrineDocument41 pagesUrineWahab KhaniNo ratings yet
- Chapter X - Mechanism of Protein MetabolismDocument30 pagesChapter X - Mechanism of Protein MetabolismAngelo AngelesNo ratings yet
- Amino Acid MetabolismDocument23 pagesAmino Acid MetabolismZ ZNo ratings yet
- Metabolism of Aminoacids 2Document68 pagesMetabolism of Aminoacids 2Mi PatelNo ratings yet
- Amino Acids Metabolism (Catabolism) and Clinical SignificantDocument33 pagesAmino Acids Metabolism (Catabolism) and Clinical SignificantBehailu TsegayeNo ratings yet
- Bio - Urea CycleDocument57 pagesBio - Urea CycleMahmoud hilmyNo ratings yet
- Protein MetaboilisamDocument18 pagesProtein MetaboilisamSumit PandyaNo ratings yet
- Amino Acid Metabolism II. Urea CycleDocument39 pagesAmino Acid Metabolism II. Urea Cycleputri jessicaNo ratings yet
- Amino Acid Catabolism-Part-1: Biochemistry For Medics - Lecture Notes Professor (DR.) Namrata ChhabraDocument43 pagesAmino Acid Catabolism-Part-1: Biochemistry For Medics - Lecture Notes Professor (DR.) Namrata Chhabrashree devNo ratings yet
- Urea Cycle and Disorders - RM - F2014Document18 pagesUrea Cycle and Disorders - RM - F2014Leon WarrenNo ratings yet
- Individual Amino Acid 2Document52 pagesIndividual Amino Acid 2Mai Elsayed OmaraNo ratings yet
- UreaDocument16 pagesUreaMuhammad AmjadNo ratings yet
- Protein Turn OverDocument29 pagesProtein Turn Overabdullah zaheerNo ratings yet
- AA Metabolism HANDOUTDocument4 pagesAA Metabolism HANDOUTnajwa hajjaliNo ratings yet
- Amino Acid CatabolismDocument19 pagesAmino Acid Catabolismwmdpr4x64fNo ratings yet
- Protein and Amino Acid MetabolismDocument52 pagesProtein and Amino Acid MetabolismRisky OpponentNo ratings yet
- Katabolisme Protein Dan Asam AmimoDocument35 pagesKatabolisme Protein Dan Asam AmimoLha YhoeLaNo ratings yet
- Urea CycleDocument39 pagesUrea Cycledrismailkm20No ratings yet
- Aminoacid MetabolismDocument22 pagesAminoacid MetabolismFazal Akbar KhaliliNo ratings yet
- Item - 11, Protein MetabolismDocument12 pagesItem - 11, Protein MetabolismSheikh FahadNo ratings yet
- 3 Metabolism of Proteins & Amino AcidsDocument79 pages3 Metabolism of Proteins & Amino AcidsYashfa YasinNo ratings yet
- Urea CycleDocument4 pagesUrea Cycletfgrn7srtqNo ratings yet
- Introduction Amino Acid Matabolism and CatabolismDocument45 pagesIntroduction Amino Acid Matabolism and CatabolismAboubakar Moalim Mahad moh'd100% (1)
- Protein MetabolismDocument182 pagesProtein MetabolismSimra ZahidNo ratings yet
- Amino Acid MetabolismDocument35 pagesAmino Acid MetabolismRajkishor YadavNo ratings yet
- Proteins and Amino Acids Metabolism: Tahun Ajar 2016/2017Document47 pagesProteins and Amino Acids Metabolism: Tahun Ajar 2016/2017Aswar AyuNo ratings yet
- Biochemistry Lecture PPT 7Document16 pagesBiochemistry Lecture PPT 7Chiranjeevi JoshiNo ratings yet
- Amino Acid & Protein MetabolismDocument59 pagesAmino Acid & Protein MetabolismAndualemNo ratings yet
- Other Amino Acids Form Succinyl CoaDocument23 pagesOther Amino Acids Form Succinyl CoanyarieNo ratings yet
- Ch18 - Amino Acid Oxidation Production of UreaDocument50 pagesCh18 - Amino Acid Oxidation Production of UreaSalma KhoirunnisaNo ratings yet
- BIO 202 Biochemistry II by Seyhun YURDUGÜL: Amino Acid Metabolism I: Amino Acid BiosynthesisDocument65 pagesBIO 202 Biochemistry II by Seyhun YURDUGÜL: Amino Acid Metabolism I: Amino Acid BiosynthesisYasin Çağrı KılıçerNo ratings yet
- Lecture 5 Protein Metabolism 1Document29 pagesLecture 5 Protein Metabolism 1Elizabeth LagunasNo ratings yet
- BP U9d Protein MetabolismDocument80 pagesBP U9d Protein MetabolismChristian Angelo AgbunagNo ratings yet
- Protein and Amino Acid MetabolismDocument32 pagesProtein and Amino Acid MetabolismVirag0% (1)
- Urea CycleDocument4 pagesUrea CycleKunal DuttaNo ratings yet
- 15BT103 Biochem-UNIT 3Document53 pages15BT103 Biochem-UNIT 3Adityanair RA1711009010128No ratings yet
- Urea CycleDocument3 pagesUrea CycleSundaralingam RajNo ratings yet
- General Protein Metabolism NUB Full 2022Document11 pagesGeneral Protein Metabolism NUB Full 2022PIH SHTNo ratings yet
- Biochemistry Week 8 Amino Acid MetabolismDocument29 pagesBiochemistry Week 8 Amino Acid MetabolismKaren 3No ratings yet
- Amino Acid MetabolismDocument81 pagesAmino Acid Metabolismcharuss.394No ratings yet
- Biochemistry: Dr. Dra. Trini Suryowati, MsDocument49 pagesBiochemistry: Dr. Dra. Trini Suryowati, MsDaud ParluhutanNo ratings yet
- Urea Cycle: Dr. Amro Yousef Al-AmlehDocument45 pagesUrea Cycle: Dr. Amro Yousef Al-AmlehDr. Amro YousefNo ratings yet
- Urea Cycle 2Document21 pagesUrea Cycle 2Akinrotimi OluwadunsinNo ratings yet
- Urea CycleDocument8 pagesUrea CycleManohar PattarNo ratings yet
- (Krebs-Henseleit Cycle) : Urea FormationDocument15 pages(Krebs-Henseleit Cycle) : Urea Formationعلي عبيد العتابيNo ratings yet
- B. Katabolisme Asam Amino-1Document19 pagesB. Katabolisme Asam Amino-1M Sifal MaulanaNo ratings yet
- PROTEIN METABOLISM Dea Farha Fira Darson FineDocument44 pagesPROTEIN METABOLISM Dea Farha Fira Darson FineFarhati Mardhiyah100% (1)
- Explain The Ff. Manifestations in The Different Cases That Are Discused BelowDocument29 pagesExplain The Ff. Manifestations in The Different Cases That Are Discused BelowRodel Paulo Tangunan GarciaNo ratings yet
- Amino Acids Metabolism Final For Pharm 2014Document57 pagesAmino Acids Metabolism Final For Pharm 2014Getu LuchesaNo ratings yet
- Catabolism of Amino Acids: DR - Ula Abbas ZekiDocument49 pagesCatabolism of Amino Acids: DR - Ula Abbas ZekinameNo ratings yet
- Protein NeedsDocument9 pagesProtein NeedsPaul BugejaNo ratings yet
- Bio-Inorganic ChemistryDocument66 pagesBio-Inorganic ChemistryPhalynxNo ratings yet
- The Effects of Creatine Ethyl Ester Supplementation Combined With Heavy Resistance Training On Body Composition, Muscle Performance, and Serum and Muscle Creatine LevelsDocument15 pagesThe Effects of Creatine Ethyl Ester Supplementation Combined With Heavy Resistance Training On Body Composition, Muscle Performance, and Serum and Muscle Creatine LevelsMarcio PertileNo ratings yet
- CC Lab 6 TransesDocument6 pagesCC Lab 6 TransesCiara PamonagNo ratings yet
- Non-Protein Nitrogenous Substances: Urea CreatineDocument2 pagesNon-Protein Nitrogenous Substances: Urea CreatineLOU BLESSY CASINONo ratings yet
- Creapure CreatineDocument4 pagesCreapure CreatineAlex RogersNo ratings yet
- How To Inhibit Myostatin For Muscle Growth and Longer LifeDocument5 pagesHow To Inhibit Myostatin For Muscle Growth and Longer Lifedarthlucasx100% (3)
- Revive Stronger Get Big Stay Lean A Comprehensive Guide To Gaining Muscle PDFDocument24 pagesRevive Stronger Get Big Stay Lean A Comprehensive Guide To Gaining Muscle PDFCyrus Dickson A CruzNo ratings yet
- Non-Protein Nitrogenous Constituents of Blood - Urea, Uric Acid EtcDocument50 pagesNon-Protein Nitrogenous Constituents of Blood - Urea, Uric Acid EtcBobskinnyNo ratings yet
- Muscle Science RoundupDocument165 pagesMuscle Science RoundupAlessandro Valentim100% (2)
- Creatine SatbilityDocument2 pagesCreatine SatbilitymonstermuddyNo ratings yet
- Biohacking LiteDocument25 pagesBiohacking Litetxboxi23No ratings yet
- Work Out: To Win BIG!Document153 pagesWork Out: To Win BIG!Alessandro ValentimNo ratings yet
- 2013-UKChO-Round 1 - tcm1Document12 pages2013-UKChO-Round 1 - tcm1vermouth020No ratings yet
- Opt' Eijnde 2001Document6 pagesOpt' Eijnde 2001Prefeitura de FloresNo ratings yet
- Challenge Yourself: Beginner Mens Challenge Preparation GuideDocument23 pagesChallenge Yourself: Beginner Mens Challenge Preparation GuideUni TabberNo ratings yet
- Supplement Education For FireDocument7 pagesSupplement Education For Fireapi-530327871No ratings yet
- Disclaimer: Provided by University of Gloucestershire Research RepositoryDocument34 pagesDisclaimer: Provided by University of Gloucestershire Research Repositorynose que no seNo ratings yet
- Muscular Development August 2015 Usapdf CompressDocument180 pagesMuscular Development August 2015 Usapdf CompressSaurav VermaNo ratings yet
- Nutrition and Hydration For HandballDocument23 pagesNutrition and Hydration For HandballGiorgianaNo ratings yet
- Nutrition Guide by Calisthenics FamilyDocument25 pagesNutrition Guide by Calisthenics FamilyTommy MartelNo ratings yet
- NPNDocument42 pagesNPNreynanrolleNo ratings yet
- Creatine: The Power Supplement?Document13 pagesCreatine: The Power Supplement?api-315070214No ratings yet
- ENERGY ZONES IN SWIMMING. Genadijus Sokolovas, Ph.D. Global Sport Technology, IncDocument6 pagesENERGY ZONES IN SWIMMING. Genadijus Sokolovas, Ph.D. Global Sport Technology, IncMateusNo ratings yet
- PretestDocument3 pagesPretestAlili DudzNo ratings yet
- Nieuw 3Document12 pagesNieuw 3Jasmijn AlmuliNo ratings yet