Download as ppt, pdf, or txt
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5820)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (845)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Readers Theater Life FlightDocument5 pagesReaders Theater Life FlightCaurrine Monsalud93% (14)
- Case Study M&SDocument3 pagesCase Study M&S68Rohan Chopra0% (1)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Oncogene EtcDocument48 pagesOncogene EtcInsyirah JohariNo ratings yet
- Apoptosis: Proliferation, Differentiation and Apoptosis Are Crucial To DevelopmentDocument21 pagesApoptosis: Proliferation, Differentiation and Apoptosis Are Crucial To DevelopmentInsyirah JohariNo ratings yet
- At The Forearm.: Biceps Tendon Supinator Pronator Teres Insertion FDS OriginDocument27 pagesAt The Forearm.: Biceps Tendon Supinator Pronator Teres Insertion FDS OriginInsyirah JohariNo ratings yet
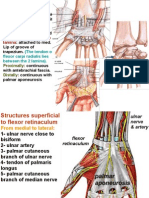
- It Enter Between Separates: Flexor RetinaculumDocument20 pagesIt Enter Between Separates: Flexor RetinaculumInsyirah JohariNo ratings yet
- Muscles of Back of ForearmDocument15 pagesMuscles of Back of ForearmInsyirah JohariNo ratings yet
- Flexor & Extensor RetinaculumDocument13 pagesFlexor & Extensor RetinaculumInsyirah Johari100% (1)
- Beginning:: It Is The Larger of Two Terminal Branches of Brachial ArteryDocument18 pagesBeginning:: It Is The Larger of Two Terminal Branches of Brachial ArteryInsyirah JohariNo ratings yet
- Fertilization: The Process by Which Male and Female Gametes Fuse To Form A Unicellular ZygoteDocument24 pagesFertilization: The Process by Which Male and Female Gametes Fuse To Form A Unicellular ZygoteInsyirah JohariNo ratings yet
- Front of Forearm LecturesDocument19 pagesFront of Forearm LecturesInsyirah JohariNo ratings yet
- 3rd Week of Embryo DevelopmentDocument20 pages3rd Week of Embryo DevelopmentInsyirah JohariNo ratings yet
- Folding, EmbryoDocument19 pagesFolding, EmbryoInsyirah JohariNo ratings yet
- Embryology: Is The Study of Human DevelopmentDocument16 pagesEmbryology: Is The Study of Human DevelopmentInsyirah Johari100% (1)
- Thorax 1Document109 pagesThorax 1Insyirah JohariNo ratings yet
- Dont Be Fuuld by JuuLDocument3 pagesDont Be Fuuld by JuuLGabriel ParksNo ratings yet
- 哈佛商业评论 (Harvard Business Review)Document7 pages哈佛商业评论 (Harvard Business Review)h68f9trq100% (1)
- Gorillas 191314 0Document2 pagesGorillas 191314 0Marisabel SernaNo ratings yet
- Ajava 6C 2160707 PDFDocument4 pagesAjava 6C 2160707 PDFanon4819No ratings yet
- Vis Tra Frost Beard The HardyDocument13 pagesVis Tra Frost Beard The HardyBrianNo ratings yet
- Clarisse Gulosino and Jonah Liebert 2012 - Review of The Way of The Future, Education Savings Accounts For Every American FamilyDocument12 pagesClarisse Gulosino and Jonah Liebert 2012 - Review of The Way of The Future, Education Savings Accounts For Every American Familyluiz carvalhoNo ratings yet
- Soal Kalimat Prohibition SMP Kelas 7Document3 pagesSoal Kalimat Prohibition SMP Kelas 7yura chanNo ratings yet
- Open-Source Software For Automated Rodent Behavioral AnalysisDocument12 pagesOpen-Source Software For Automated Rodent Behavioral AnalysisGonzalo OrtegaNo ratings yet
- Pantone PDFDocument55 pagesPantone PDFMj Jack Rachmanto100% (1)
- Full Download pdf of (eBook PDF) The Global Casino: An Introduction to Environmental Issues 6th Edition all chapterDocument43 pagesFull Download pdf of (eBook PDF) The Global Casino: An Introduction to Environmental Issues 6th Edition all chapternahoonabid100% (5)
- The Elusive Butterfly: Groove..Document5 pagesThe Elusive Butterfly: Groove..Nasciturus666No ratings yet
- Mole Concept DPP-2 - 501352Document1 pageMole Concept DPP-2 - 501352Vatsal BhargavaNo ratings yet
- Adonis SaturnoDocument6 pagesAdonis SaturnoRaymond G. PanhonNo ratings yet
- 10 Female Superheroes Who Depict Women EmpowermentDocument3 pages10 Female Superheroes Who Depict Women EmpowermentSharjeel ZamanNo ratings yet
- Heatmaster - Hot Water BoilerDocument1 pageHeatmaster - Hot Water BoilerNorbertoNo ratings yet
- International Business: Case Study-Report-3 18-02-2022Document5 pagesInternational Business: Case Study-Report-3 18-02-2022swapnil anandNo ratings yet
- Fuji ManualDocument166 pagesFuji Manualasbel gamNo ratings yet
- DLL All Subjects 2 q2 w9 d1Document9 pagesDLL All Subjects 2 q2 w9 d1Nicole AragonNo ratings yet
- Phlebotomist TrackerDocument62 pagesPhlebotomist Trackertarun sharmaNo ratings yet
- Introduction To Business ResearchDocument23 pagesIntroduction To Business ResearchKhalid ElGhazouliNo ratings yet
- BBPS Kennedy03 PDFDocument8 pagesBBPS Kennedy03 PDFCyz RobertNo ratings yet
- OceanofPDF - Com Return To The Carnival of Horrors - RL StineDocument170 pagesOceanofPDF - Com Return To The Carnival of Horrors - RL Stinedinulaka kaluthanthireeNo ratings yet
- RahmaDocument3 pagesRahmaAyyub RamadanNo ratings yet
- 2018 Bibliography I. Calvin's Life and TimesDocument25 pages2018 Bibliography I. Calvin's Life and Timesmoreirajanne545No ratings yet
- COP2800 MidtermStudyGuideDocument31 pagesCOP2800 MidtermStudyGuideCatherine CrutcherNo ratings yet
- Multimodal Transport Law - The Law Applicable To Multimodal Contract For The Carriage of Goods (PDFDrive)Document561 pagesMultimodal Transport Law - The Law Applicable To Multimodal Contract For The Carriage of Goods (PDFDrive)Isaac Joshua AganonNo ratings yet
- Project Report Dam Level WarningDocument8 pagesProject Report Dam Level Warningpankaj_yadav007No ratings yet
- Rural - India Oct 10 EDELWEISSDocument89 pagesRural - India Oct 10 EDELWEISSpkn04100% (1)