Download as pptx, pdf, or txt
You might also like
- Merloni Case - Group 1Document11 pagesMerloni Case - Group 1Prasanta Mondal100% (2)
- Item Specification RemarkDocument10 pagesItem Specification RemarkKada Ben youcefNo ratings yet
- Blood Vessel Diseases LEVEL IIDocument33 pagesBlood Vessel Diseases LEVEL IIDavid Ogechi AtandiNo ratings yet
- Cutaneous Vasculitis: Oktay Taşkapan, MDDocument34 pagesCutaneous Vasculitis: Oktay Taşkapan, MDKada Ben youcefNo ratings yet
- VasculitisDocument61 pagesVasculitisshadabNo ratings yet
- L27 VasculitisDocument28 pagesL27 VasculitisGazarNo ratings yet
- Vasculitis 1Document47 pagesVasculitis 1Rusty RyanNo ratings yet
- VasculitisDocument63 pagesVasculitisPutu 'yayuk' Widyani WiradiraniNo ratings yet
- Cvs Lec 2Document64 pagesCvs Lec 2data4fourgNo ratings yet
- Prinsip Gangguan Musculoskeletal: Inflamasi,, Acute and Chronic Arthritis,, OsteoporosisDocument110 pagesPrinsip Gangguan Musculoskeletal: Inflamasi,, Acute and Chronic Arthritis,, OsteoporosisSepti Fadhilah SPNo ratings yet
- Chapter 11 - Blood VesselsDocument6 pagesChapter 11 - Blood VesselsAgnieszka WisniewskaNo ratings yet
- Vaskulitis-1Document38 pagesVaskulitis-1LIEBERKHUNNo ratings yet
- Ishihara Test PlatesDocument21 pagesIshihara Test PlatesAde Ayuningsih UtamiNo ratings yet
- EL CP 33-34 - CollagenosesDocument155 pagesEL CP 33-34 - CollagenosesUmar RahmanNo ratings yet
- WBC 1Document74 pagesWBC 1Johnmerson YapNo ratings yet
- VasculittisDocument28 pagesVasculittisBernamai CalamNo ratings yet
- Routine Blood Examination For InfectionDocument43 pagesRoutine Blood Examination For InfectionElmyLiantika MaranantanNo ratings yet
- Cns Infection 3rd Year MBBSDocument66 pagesCns Infection 3rd Year MBBSKritick BhandariNo ratings yet
- Rheumatic Heart Disease: Dr.K.Sathish Kumar, MD (Hom) .Document14 pagesRheumatic Heart Disease: Dr.K.Sathish Kumar, MD (Hom) .mnr hmcNo ratings yet
- AIBD, Vasculitis and PanniculitisDocument9 pagesAIBD, Vasculitis and PanniculitisSudesna Roy ChowdhuryNo ratings yet
- 1. CVS خديجهDocument19 pages1. CVS خديجهsafsafpath7No ratings yet
- Leukocytes: - GranulocytesDocument38 pagesLeukocytes: - GranulocytesCurrencyNo ratings yet
- Disseminated Intravascular CoagulopathyDocument15 pagesDisseminated Intravascular Coagulopathyapi-26826496No ratings yet
- Pathology AssignmentDocument15 pagesPathology AssignmentsumairaNo ratings yet
- Vasculitis ReviewDocument132 pagesVasculitis Reviewajmal_rashid@hotmail.com100% (2)
- Anak 3.1 Infective Endocarditis DRTLTDocument21 pagesAnak 3.1 Infective Endocarditis DRTLTAnastasia PinkyNo ratings yet
- Inflammation Bvsc3 DR Grace Lecture NotesDocument154 pagesInflammation Bvsc3 DR Grace Lecture NotesnyarieNo ratings yet
- Pediatric Systemic Lupus Erythematosus: Pediatric Rheumatology Red Team Resident Teaching SeriesDocument52 pagesPediatric Systemic Lupus Erythematosus: Pediatric Rheumatology Red Team Resident Teaching SeriesLili ManaoNo ratings yet
- Clinico Pathological ChartsDocument132 pagesClinico Pathological Chartsomatre210No ratings yet
- Vasculitis: Anapi. Aragon. de Leon. SamsonDocument16 pagesVasculitis: Anapi. Aragon. de Leon. SamsonGeramyl Ramos AnapiNo ratings yet
- Bleeding Disorders 1Document152 pagesBleeding Disorders 1Shameena KnNo ratings yet
- Rheumatic Valvular DiseaseDocument28 pagesRheumatic Valvular DiseaseBenallouaminaNo ratings yet
- Systemic Vasculitis: A Clinical Approach: Geordie Lawry MDDocument65 pagesSystemic Vasculitis: A Clinical Approach: Geordie Lawry MDLydwiNa Jc100% (1)
- Menigitis EncephalitisDocument63 pagesMenigitis EncephalitisHussain Azhar100% (1)
- PsoriasisDocument32 pagesPsoriasisEffah FestusNo ratings yet
- SarcoidosisDocument95 pagesSarcoidosisvnybhagat100% (2)
- GlomerulonephritisTips and New Pathts UCMI (1)Document30 pagesGlomerulonephritisTips and New Pathts UCMI (1)Afonso Henriques NunesNo ratings yet
- Atherosclerosis PresentationDocument90 pagesAtherosclerosis PresentationAbu SaifNo ratings yet
- WBCDocument25 pagesWBCcelecosibNo ratings yet
- BrucellosisDocument38 pagesBrucellosisEslam HamadaNo ratings yet
- Gram-Positive Cocci 2Document45 pagesGram-Positive Cocci 2Iftikhar AhmadNo ratings yet
- Diseases of Blood and Blood Forming OrgansDocument19 pagesDiseases of Blood and Blood Forming OrgansAMIT GUPTANo ratings yet
- Systemic SclerosisDocument55 pagesSystemic Sclerosisnainnu81No ratings yet
- Smle Pictorial JunayedDocument86 pagesSmle Pictorial Junayedmonirul islam shohan100% (1)
- Non-Neoplastic Disorders: Lymphoid SystemDocument36 pagesNon-Neoplastic Disorders: Lymphoid SystemMutiana Muspita JeliNo ratings yet
- Leukopenia and Bone Marrow TransplantationDocument20 pagesLeukopenia and Bone Marrow Transplantationdhanya jayanNo ratings yet
- Family NeisseriaDocument38 pagesFamily NeisseriakavyaNo ratings yet
- (K25) Path - Musculosceletal FK Part 2Document69 pages(K25) Path - Musculosceletal FK Part 2Virginia JawaNo ratings yet
- Connective Tissue Disorders: DR Josephine Ojoo MBCHB FRCP CCST (Resp) Dip Hiv Med Senior Lecturer Maseno UniversityDocument70 pagesConnective Tissue Disorders: DR Josephine Ojoo MBCHB FRCP CCST (Resp) Dip Hiv Med Senior Lecturer Maseno UniversityMalueth AnguiNo ratings yet
- Chapter 2 - Acute and Chronic InflammationDocument12 pagesChapter 2 - Acute and Chronic InflammationAgnieszka WisniewskaNo ratings yet
- Cardiac Pathology 2 Alzahrawi PDFDocument43 pagesCardiac Pathology 2 Alzahrawi PDFMΘΘNNo ratings yet
- 20 Bacterial MeningitisDocument35 pages20 Bacterial MeningitisBhakti WashilkarNo ratings yet
- WSU Renal ReviewDocument100 pagesWSU Renal ReviewLeticia BornsteinNo ratings yet
- Rheumatic Fever & Infective EndocarditisDocument46 pagesRheumatic Fever & Infective EndocarditisMosab MasoudNo ratings yet
- Connective Tissue DiseasesDocument22 pagesConnective Tissue DiseasesMohd Johari Mohd ShafuwanNo ratings yet
- Schistosomiasis: Presenter Hizkiel Abera (MD, Imr1)Document40 pagesSchistosomiasis: Presenter Hizkiel Abera (MD, Imr1)BethelAberaHaydamoNo ratings yet
- Wa0007.Document33 pagesWa0007.Rigzemo ZaidNo ratings yet
- EM Acute Inflammation SBDocument33 pagesEM Acute Inflammation SBPratya DeyNo ratings yet
- Approach To Systemic Vasculitis - Print VersionDocument32 pagesApproach To Systemic Vasculitis - Print Versionnamhom.md44No ratings yet
- Histopathology of Kidney: Krisna MurtiDocument58 pagesHistopathology of Kidney: Krisna MurtiYUFFANo ratings yet
- Engine Data ListDocument45 pagesEngine Data ListKada Ben youcefNo ratings yet
- Component Item SpecificationDocument22 pagesComponent Item SpecificationKada Ben youcefNo ratings yet
- C200 - Pre-Heating SystemDocument7 pagesC200 - Pre-Heating SystemKada Ben youcefNo ratings yet
- C200 - CDPF SystemDocument12 pagesC200 - CDPF SystemKada Ben youcefNo ratings yet
- C200 - Intake SystemDocument9 pagesC200 - Intake SystemKada Ben youcefNo ratings yet
- 20 Cat Devant Des Douleurs Abdominales de L'enfantDocument51 pages20 Cat Devant Des Douleurs Abdominales de L'enfantKada Ben youcefNo ratings yet
- C200 - Cooling SystemDocument11 pagesC200 - Cooling SystemKada Ben youcefNo ratings yet
- C200 - Starting SystemDocument5 pagesC200 - Starting SystemKada Ben youcefNo ratings yet
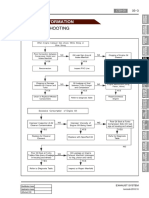
- Troubleshooting: 1) Work FlowDocument4 pagesTroubleshooting: 1) Work FlowKada Ben youcefNo ratings yet
- C200 - Fuel SystemDocument27 pagesC200 - Fuel SystemKada Ben youcefNo ratings yet
- C200 - Engine GeneralDocument16 pagesC200 - Engine GeneralKada Ben youcefNo ratings yet
- C200 - Lubrication SystemDocument5 pagesC200 - Lubrication SystemKada Ben youcefNo ratings yet
- D20Dtf Engine: 1. SpecificationDocument26 pagesD20Dtf Engine: 1. SpecificationKada Ben youcefNo ratings yet
- Dsi 6 Speed Auto TransaxleDocument24 pagesDsi 6 Speed Auto TransaxleKada Ben youcefNo ratings yet
- C200 - Cruise Control SystemDocument12 pagesC200 - Cruise Control SystemKada Ben youcefNo ratings yet
- Drive Shaft and AxleDocument4 pagesDrive Shaft and AxleKada Ben youcefNo ratings yet
- Unit Description SpecificationDocument13 pagesUnit Description SpecificationKada Ben youcef100% (1)
- General Information ChassisDocument28 pagesGeneral Information ChassisKada Ben youcefNo ratings yet
- Specification: Unit ConstructionDocument7 pagesSpecification: Unit ConstructionKada Ben youcefNo ratings yet
- C200 - E-Egr SystemDocument9 pagesC200 - E-Egr SystemKada Ben youcefNo ratings yet
- Specification: 1) Appearance of WheelsDocument6 pagesSpecification: 1) Appearance of WheelsKada Ben youcefNo ratings yet
- C200 - Charge SystemDocument10 pagesC200 - Charge SystemKada Ben youcefNo ratings yet
- Suspension SystemDocument13 pagesSuspension SystemKada Ben youcefNo ratings yet
- Power Steering SystemDocument5 pagesPower Steering SystemKada Ben youcefNo ratings yet
- Propeller Shaft (4Wd Only) : 1. SpecificationDocument3 pagesPropeller Shaft (4Wd Only) : 1. SpecificationKada Ben youcefNo ratings yet
- Specification: Unit Description Specification System OperationDocument4 pagesSpecification: Unit Description Specification System OperationKada Ben youcefNo ratings yet
- Description SpecificationDocument7 pagesDescription SpecificationKada Ben youcefNo ratings yet
- System Overview: 1) Terms and DefinitionDocument9 pagesSystem Overview: 1) Terms and DefinitionKada Ben youcefNo ratings yet
- Specification: Unit Description Specification ABS ESP HecuDocument22 pagesSpecification: Unit Description Specification ABS ESP HecuKada Ben youcefNo ratings yet
- Macroeconomics 1Document25 pagesMacroeconomics 1eunicemaraNo ratings yet
- Sancho Agapito Jackelin CarolDocument36 pagesSancho Agapito Jackelin CarolTeòfilo Quispe MedinaNo ratings yet
- Presentation Sussex Conference (1st Year PHD Student)Document9 pagesPresentation Sussex Conference (1st Year PHD Student)vsavvidouNo ratings yet
- Aldo Colombini - Pre-DeckabilityDocument4 pagesAldo Colombini - Pre-DeckabilityRoberto GuerínNo ratings yet
- Report On Caste Based Atrocities On Dalits in Varanasi and Surrounding Areas in Uttar PradeshDocument31 pagesReport On Caste Based Atrocities On Dalits in Varanasi and Surrounding Areas in Uttar PradeshPeoples' Vigilance Committee on Human rightsNo ratings yet
- Direct & Indirect SpeechDocument29 pagesDirect & Indirect SpeechHumanist English ManNo ratings yet
- Child Adolescent Module5 Factors Affecting Cognitive DevelopmentDocument15 pagesChild Adolescent Module5 Factors Affecting Cognitive DevelopmentBunso A. Loresto60% (5)
- Aries (Astrology) From Wikipedia, The Free EncyclopediaDocument5 pagesAries (Astrology) From Wikipedia, The Free Encyclopediablessed cccNo ratings yet
- Vintage Airplane - Feb 1987Document32 pagesVintage Airplane - Feb 1987Aviation/Space History LibraryNo ratings yet
- SAP Contoh SoalDocument28 pagesSAP Contoh SoalArifin SantosoNo ratings yet
- A Summer Training Project Report ON: Iftm University, MoradabadDocument6 pagesA Summer Training Project Report ON: Iftm University, MoradabadbuddysmbdNo ratings yet
- Lab Test HivDocument2 pagesLab Test HivCindy MoraNo ratings yet
- DLP Entrep Revised 102Document1 pageDLP Entrep Revised 102ClintNo ratings yet
- Unit 11.conditionals. ExercisesDocument14 pagesUnit 11.conditionals. Exerciseslondonfaidel6511No ratings yet
- Kokomo Blighted HomesDocument9 pagesKokomo Blighted HomesKyle BloydNo ratings yet
- Sustainability 13 10705 v2 PDFDocument18 pagesSustainability 13 10705 v2 PDFVũ Ngọc Minh ThuNo ratings yet
- BCB StainingDocument10 pagesBCB StainingAmbikaprasanna SahaNo ratings yet
- 1111Document9 pages1111trungtroangNo ratings yet
- 1-112 Attack Helicopter ManualDocument351 pages1-112 Attack Helicopter ManualParijata JATA Mackey100% (3)
- Festo GrafcetDocument34 pagesFesto GrafcetSertug Başar50% (2)
- Accomplishment Report BrigadaDocument2 pagesAccomplishment Report BrigadaVincent rexie AsuncionNo ratings yet
- CO4CRT12 - Quantitative Techniques For Business - II (T)Document4 pagesCO4CRT12 - Quantitative Techniques For Business - II (T)Ann Maria GeorgeNo ratings yet
- Kilosbayan Inc. vs. GuingonaDocument1 pageKilosbayan Inc. vs. GuingonaJo LumbresNo ratings yet
- 4302 M.Sc. OBSTETRIC AND GYNAECOLOGICAL NURSINGDocument89 pages4302 M.Sc. OBSTETRIC AND GYNAECOLOGICAL NURSINGSteny Ann VargheseNo ratings yet
- Monthly Statement: This Month's SummaryDocument10 pagesMonthly Statement: This Month's SummaryPritam JanaNo ratings yet
- Application & Technical Manual 02 Structural SteelDocument65 pagesApplication & Technical Manual 02 Structural SteelTian NgNo ratings yet
- Exercise 09 - Investigating HazardsDocument23 pagesExercise 09 - Investigating HazardsJill ClarkNo ratings yet
- JohnsonEvinrude ElectricalDocument5 pagesJohnsonEvinrude Electricalwguenon100% (1)
- Angular Cheilitis - Causes, Symptoms, Treatment and MoreDocument7 pagesAngular Cheilitis - Causes, Symptoms, Treatment and Moremefav7778520No ratings yet